Farmacocinetica studio dei fattori che determinano la quantità di farmaco presente nei siti d'azione - Università degli Studi di Ferrara
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Farmacocinetica
studio dei fattori che determinano la quantità di
farmaco presente nei siti d’azione
Assorbimento
Distribuzione
Metabolismo (biotrasformazione)
Eliminazione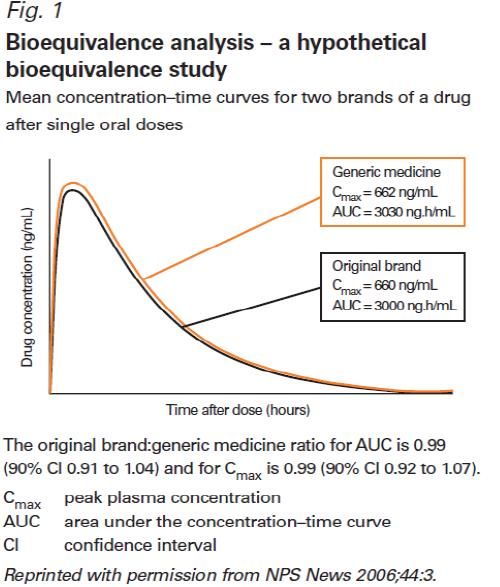
…da non confondere con la Farmacodinamica che invece si occupa dello studio dei meccanismi d’azione dei farmaci e del loro impatto sulla materia vivente
FARMACO SOMMINISTRATO
(A) assorbimento
(C) attivazione/ (D) escrezione/
inattivazione metabolica riassorbimento
pool di farmaco disponibile
e/o di metabolita attivo
nell’organismo
(B) distribuzione
FARMACO O METABOLITA
ATTIVO AL SITO D’AZIONE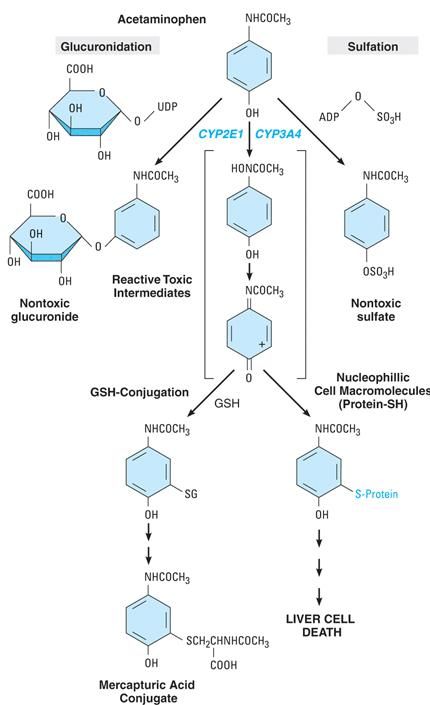
Il farmaco deve attraversare le membrane
biologiche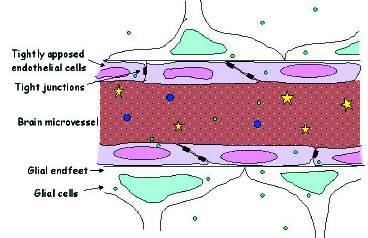
• Le caratteristiche di un farmaco predittive della
sua distribuzione e biodisponibilità a livello dei
siti d’azione sono:
– Peso molecolare e caratteristiche strutturali
– Grado di ionizzazione
– Liposolubilità relativa delle forme ionizzate e non
ionizzate
– Legame alle prot plasmatiche e tissutali
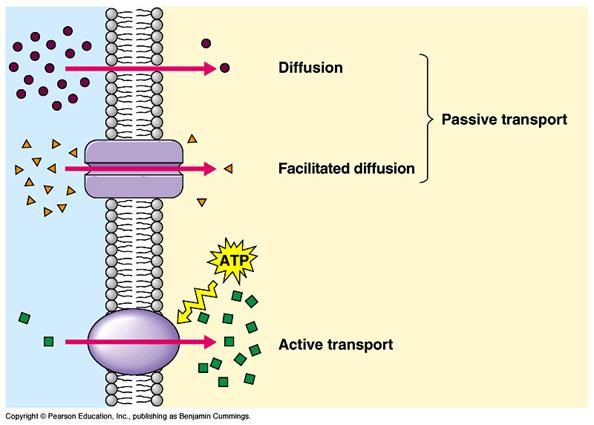
• Il passaggio delle membrane avviene per processi
passivi ed attivi• Il passaggio delle membrane può avvenire per
• Flusso passivo
– trasporto paracellulare (molecole basso PM)
– Diffusione passiva (la >parte dei ff)
• Trasportatore
– trasporto attivo, richiede energia
• Primario, accoppiato direttamente all’idrolisi di ATP (es MDR)
• Secondario Es sinporti o antiporti
(ass glucosio m GI e renali via SGLT1 e SGLT2,)
– diffusione facilitata (es GLUT4 nel muscolo, glicoproteina-
P), non richiede energia
• Fago/pinocitosi
– Grosse molecole proteicheTrasporto attivo
Mediato da trasportatori e caratterizzato da:
Richiesta di energia
Movimento contro gradiente ec
Saturazione
Selettività
Inibizione competitiva da parte di
composti co-trasportatiENDOCITOSI •pinocitosi (liquidi) •fagocitosi (solidi)
• FATTORI CHE REGOLANO LA VELOCITA’ DI PASSAGGIO
DI UN FARMACO ATTRAVERSO LE MEMBRANE
• DIFFUSIBILITA’ (LEGGE DI FICK)
• DISSOCIABILITA' (eq. HENDERSON-HASSELBACH)
• COEFFICIENTE DI RIPARTIZIONE
•
• LEGGE DI FICK
• Vel diffusione Dm/Dt = -DA * dc/dx
– A= area di applicazione
– dc= gradiente di concentrazione
– dx= spessore della membrana
– D= costante di permeabilità (coeff di diffusione)
• Dipende dalle prop della molecola (es lipofilia, PM)DISSOCIABILITA’
Molti farmaci sono acidi o basi deboli, presenti in soluzione in
forma ionizzata/non ionizzata
in genere le forme NON ionizzate sono più liposolubili e
diffondono più facilmente
EQUAZIONE DI HENDERSON-HASSELBACH
[A-] [H+] 1
Ka = pKa = log
[AH] Ka
[Diss-]
Per acidi deboli: pH-pKa = log[Indiss]
[Indiss]
Per basi deboli: pH-pKa = log [Diss]
Il pKa (costante di dissociazione) è il valore di pH alla
quale metà del farmaco si trova in forma ionizzataprincipio dell’ «intrappolamento ionico» • Anti-H1 di II generazione: sono protonati a pH fisiologico perciò non passano la BEE • Eliminazione renale dei ff è modificata dalla variazione del pH urine: – diuresi forzata alcalina – diuresi forzata acida • Il plasma fetale è più acido (7-7.2) rispetto a ql materno (7.4). Ff basici vengono intrappolati
Farmacocinetica: assorbimento • Movimento del farmaco dal sito di somministrazione/applicazione al compartimento centrale • L’entità e la velocità dell’assorbimento variano a seconda della via di somministrazione • Biodisponibilità: frazione della quantità di farmaco somministrata che raggiunge il sito d’azione o il fluido biologico dal quale il farmaco raggiunge il sito d’azione
Tmax
La biodisponibilità si misura
determinando la concentrazione
plasmatica di farmaco in relazione
Cmax al tempo.
Viene quantificata sulla base della
AUC AUC («area under the curve»)
• Biodisponibilità assoluta
AUC x /AUV ev
Rapporto tra AUC per una stessa dose somministrata per determinata via
di somm e per via endovenosa
• Biodisponibilità relativa
Rapporto tra AUC ottenute con diverse formulazioni della stessa dose di
farmacoDue farmaci si dicono bioequivalenti quando AUC, Cmax e Tmax sono sostanzialmente sovrapponibili, con un margine di tolleranza del 20%. Due formulazioni sono definite bioequivalenti quando la differenza tra le loro biodisponibilità rientri in un intervallo predefinito come "intervallo accettabile" di bioequivalenza, fissato nell’intervallo 0,80-1,25 convenzionalmente ritenuto compatibile con l'equivalenza terapeutica. Il margine si riduce a 0,9-1,11 per farmaci a basso IT (es antiepilettici, warfarin, antiaritmici, TCA,…)
Farmaco generico • Medicinale che ha la stessa composizione qualitativa e quantitativa di sostanze attive, la stessa forma farmaceutica, la stessa modalità di rilascio del medicinale di riferimento o «originator» (equivalente farmaceutico) ma che ha anche una bioequivalenza con il medicinale di riferimento, dimostrata da studi appropriati di biodisponibilità • Il termine «farmaco generico» (1995) è stato quindi sostituito (2005) da «farmaco equivalente» • Due farmaci equivalenti hanno la stessa equivalenza terapeutica
• FATTORI CHE REGOLANO LA VELOCITA' DI ASSORBIMENTO • VARIABILI DIPENDENTI DAL FARMACO – COEFFICIENTE DI RIPARTIZIONE – COEFFICIENTE DI DIFFUSIONE – DISSOCIABILITA' • VARIABILI DIPENDENTI DALLA FORMA FARMACEUTICA – FORME SOLIDE vs LIQUIDE (disgregabilità della compressa) – IMPORTANZA DEL SOLVENTE (es. preparazioni ritardo) • VARIABILI DIPENDENTI DALLA SUPERFICIE ASSORBENTE – ESTENSIONE – PERMEABILITA' – VASCOLARIZZAZIONE – pH
• pH di alcune superficie di assorbimento – Stomaco 1-5 – Duodeno 5-8 – Digiuno 7-8 – Colon 7-7.5 – Congiuntiva 7.2-8 – Cute 4.2-6.5
Via orale
• Metodo più comune. Ma anche più
economico, sicuro e comodo
• Svantaggi:
– Ass limitato di alcuni ff per caratt chim-fisiche
– Emesi
– Distruzione per enzimi digestivi o pH (idrolisi)
– Irregolarità per interferenza con cibo, velocità
svuotam gastrico, altri ff
– Compliance
– Metabolismo (mucosa GI, fegato, flora batterica)• Il passaggio delle membrane GI può avvenire per • Flusso passivo – trasporto paracellulare (molecole basso PM) – Diffusione passiva (la >parte dei ff) • Trasportatore – trasporto attivo, richiede energia – diffusione facilitata, non richiede energia • Fago/pinocitosi – Grosse molecole proteiche • Il passaggio delle membrane GI è bidirezionale in quanto esistono sistemi di estrusione attiva, mediati da carriers (es glicoproteina-P)
• FATTORI CHE REGOLANO L'ASSORBIMENTO INTESTINALE
• CARATT FISICO-CHIMICHE DEL FARMACO
• PERFUSIONE E AREA
– (stomaco 0,15 Lt/min; 1 mq)
– (int tenue 1 Lt/min; 40 mq)
• VELOCITA' DI SVUOTAMENTO GASTRICO
(metoclopramide e ass. di digossina)
• VELOCITA' DI TRANSITO
(es. lassativi nelle intossicazioni)
• INTERAZIONI COL CIBO O ALTRE SOSTANZE
(es. AMINOACIDI E l-DOPA, ES LATTICINI E TETRACICLINE)
• EFFETTO DI PRIMO PASSAGGIO
• EFFETTO DEL pH GASTRICO• Preparazioni a rilascio controllato: forniscono
un ass lento e costante nel tempo (> 8 ore)
• Vantaggi:
– effetto prolungato (utile per ff ad emivita < 4ore)
– Riduzione frequenza somm (migliora la
compliance)
– Copertura notturna
– Riduzione picchi di farmaco (ed eff collaterali)• EFFETTO DELLA FLORA BATTERICA INTESTINALE • • 1. reazioni di RIDUZIONE • riduzione dei nitrati a nitriti con rischio di metaHb (imp nei bambini dove la crescita di E. Coli è favorita dal maggiore pH gastrico) • • 2. reazioni di NITROSAZIONE • (nitriti-nei conservanti- e ammine II-nei pesci, vegetali- reagiscono • con formazione di nitrosammine cancerogene) • • 3. reazioni BETA-GLICURONIDASICHE • (circolo entero-epatico)
• Somm sublinguale
– Rilevante per alcuni farmaci (es nitroglicerina)
– Evita il metabolismo di primo passaggio (circolaz
venosa orale v. cava superiore)
• Somm rettale: vantaggiosa in qualche caso:
– Circa il 50 % del farmaco assorbito nel retto non
passa dal fegato (minore eff primo pass)
– Il CYP3A4 non viene espresso dalle cell rettali
– Svantaggi: ass irregolare, irritazioni della mucosaAssorbimento transdermico.
L’epidermide si comporta da barriera lipidica, il derma è molto
permeabile
Strati della pelle umana
A = ghiandola sudoripara
B = follicolo pilifero
C = ghiandola sebacea
ogni annesso cutaneo ha la propria
irrorazione• Dipende da: – Caratt del farmaco (liposolubilità) – Estensione e zona – Stato di integrità, infiammazione, vascolarizz – Stato di idratazione (bendaggio occlusivo) • Cerotti medicati a base di nicotina, scopolamina, nitroglicerina, testosterone ed estrogeni, analgesici • Sufficiente dare tox (es insetticidi inib AChE, allergie a FANS)
Applicazioni topiche • Mucose: • Scopo: ottenere effetti locali. Es congiuntiva, rino/orofaringe, vagina, colon, uretra, vescica. • Ma anche per effetti sistemici (es applicazioni intranasali di ormoni, es appl vaginali di contraccettivi, es gomme alla nicotina) • Occhio: • Scopo: effetti locali (assorbimento dopo passaggio della cornea) • Possono insorgere effetti sistemici (drenaggio nasolacrimale) es steroidi, beta-bloccanti
Vie parenterali
endovenosa, intramuscolare, sottocutanea, intrarteriosa, epidurale/intratecale
• Vantaggi:
• biodisponibilità rapida, ampia e prevedibile
• Condizioni di emergenza con pz non cooperanti
• Necessarie con alcuni farmaci, non somm per os
• Svantaggi:
– Mantenere asepsi (es ev lente, o it)
– Dolore
– Spesso richiede un operatore
•
• L’assorbimento avviene per diffusione semplice, secondo gradiente
• Il processo è limitato dall’estensione dei superfici capillari (gli endoteli
sono poco selettivi) e dalla solubilità del ff nei liquidi interstiziali
• Le proteine vengono ass più lentamente, passando per la via linfatica
• I ff somm per via sistemica sono soggetti all’effetto di primo passaggio
polmonare, che funge anche da filtro per fini particelle in sospensione• Via endovenosa
• Vantaggi:
– rapida e completa biodisponibilità.
– Monitorare la risposta clinica (es anestesia)
– Possibile iniettare anche soluz irritanti (il sangue
diluisce)
• Svantaggi:
– Reaz indesiderate (es sincope)
– Sotto controllo di un operatore
– No ff in veicoli oleosi, o ff che causano precipitaz
degli elementi corpuscolati del sangue, o danno
anemia emolitica• Via sottocutanea
• Utilizzata solo per ff che non danno irritazione
tissutale (necrosi, dolore, distacco tissutale)
• Assorbimento modificabile
– es insulina con azione rapida, intermedia o lunga)
– Es anestetico locale (lidocaina) + vasocostrittore
(adrenalina)
– Dispositivi contraccettivi a lunga durata d’azione
(impianti di etonorgestrel; implanon®)• Via intramuscolare • Ass rapido di ff in sol acquosa. • Ma anche veicoli oleosi per prep deposito (depot) che garantiscono un ass lento e costante • Ass dipende dalla vascolarizzazione, che può essere incrementata da esercizio fisico o frizione. • Ass deltoide > grande gluteo • Ass nel gluteo: gluteo uomini > donne • Via intrarteriosa • Via intratecale: nel liquor, nello spazio compreso tra pia madre e aracnoide (es analgesia con oppioidi, infezioni SNC) • Via epidurale (peridurale): tra la dura madre e le ossa, es analgesia parto od operatoria).
Lo spazio epidurale
Dura madre
nervi
Midollo spinale liquorVia polmonare • Rapido, molto efficiente, non effetto di primo passaggio epatico • Gli alveoli superficie di ass ideale: – AREA ALVEOLI 100mq – AREA CAPILLARI 140mq – FLUSSO EMATICO 90ml/sec Farmacologia: gas anestetici, farmaci nebulizzati (aerosol) per terapia locale (es antiasmatici) Tossicologia (es particolato; sostanze d’abuso)
• GAS E VAPORI • FATTORI CHE CONDIZIONANO L'ASSORBIMENTO: – FILTRO NASALE (solo se il gas è molto reattivo e idrosolubile) – IDROSOLUBILITA' (meno import la liposolubilità e il grado di ionizzazione) – REATTIVITA' TISSUTALE (es. CO ed ERITROCITI) – COEFF RIPART SANGUE/GAS • L'ASSORBIMENTO DEL GAS AVVIENE PER DISSOLUZIONE SECONDO LA LEGGE DI HENRY. ALL'EQUILIBRIO LA QUANTITA' ASSORBITA SARA DETTATA DAL COEFF RIP SANGUE/GAS. • ETILENE (BASSO RAPPORTO= 0.14). SATURAZIONE A BASSE CONC (tempo di equilibrio 8-21 min). • SE NE SCIOGLIE POCO E QS % PUO' ESSERE INCREMENTATA AUMENTANDO IL FLUSSO EMATICO (LIMITE DI PERFUSIONE) • • CLOROFORMIO (RAPPORTO=15). SATURAZIONE AD ALTE CONC (tempo di equilibrio 1 ora e più). • LA QUOTA ASSORBITA PUO’ ESSERE INCREMENTATA AUMENTANDO LA VENTILAZIONE (LIMITE DI VENTILAZIONE).
• AEROSOL E PARTICELLE
• FATTORI CHE CONDIZIONANO L'ASSORBIMENTO:
– IDROSOLUBILITA'
– TIPO DI AEROSOL
• DIMENSIONI
• > 5 uM NASOFARINGE
• 2-5 uM REGIONI TRACHEOBRONCHIALI
• 1 uM ALVEOLI (ASSORBIMENTO)
• MECCANISMI DI RIMOZIONE
– Clearance muco ciliare
– FAGOCITOSI (MACROFAGI) ALVEOLARE (ASBESTO, SILICE)
– RIMOZIONE PER VIA LINFATICA (sop. proteine)
• l'endotelio che ricopre i capillari linfatici è permeabile a mol PM> 20.000Distribuzione
Dopo l’assorbimento (o la diretta immissione
in circolo) il farmaco si distribuisce negli spazi
interstiziali e nei fluidi extracellulari• Fattori che influenzano la distribuzione:
1. CARATT FISICO-CHIMICHE DEL COMPOSTO
– diffusibilità
– dissociabilità
– coeff ripartizione
2. PERFUSIONE REGIONALE
• La prima fase della distribuzione è determinata da gittata cardiaca, volume
tissutale e vascolarizzazione
– rene, fegato 20-25 % gittata (ciascuno)
– Cervello 12% gittata
– Pelle, t. adiposo 6-10 %
• La seconda fase (REDISTRIBUZIONE) (da minuti a ore) è legata alla fase
extravascolare e a fattori di affinità
• La ripartizione sangue/tessuto è condizionata essenzialmente dal legame
alle prot plasmatiche e alla affinità del farmaco per i costituenti cellulari di
quello specifico tessuto
• La differenza di pH tra sangue e tessuto è minima (7.4 vs 7.0).
• Solo il farmaco in forma libera si distribuisce ai tessutiRIDISTRIBUZIONE
DISTRIBUZIONE DEL PENTOTHAL dopo iniezione e.v. nel cane
concentrazione concentrazione
TESSUTO (µg/g) 5 min dopo (µg/g) 60 min dopo
la somministrazione la somministrazione
plasma 15 5
liquor 3 1
fegato 30 20
cervello 24 7
rene 18 15
tessuto 4 40
adiposo3. LEGAME ALLE PROTEINE PLASMATICHE
– processo reversibile e saturabile, non lineare
– Influenzato da patologie (es ipoalbuminemie da epatopatie)
– Influenzato dalla gravidanza (es T4)
• albumina principale proteina per i ff acidi
• a1-glicoproteina acida lega i ff basici
• proteine specifiche:
– Globulina legante il testosterone o gli estrogeni
– Globulina legante la tiroxina
• FATTORI CHE REGOLANO IL LEGAME ALLE PROTEINE PLASMATICHE
• CONCENTRAZIONE DEL FARMACO (il rapporto vale alle conc
terapeutiche)
• COSTANTE DI DISSOCIAZIONE (inversamente proporzionale
all’affinità)
• NUMERO DI SITI DI LEGAME
• INTERAZIONE CON ALTRI FARMACI (SPIAZZAMENTO)• CONSEGUENZE DEL LEGAME ALLE PROTEINE PLASMATICHE
• SEQUESTRO DEL TOSSICO NEL COMPARTIMENTO EMATICO
– RIDOTTO TRASFERIMENTO NEI COMPARTIMENTI EXTRAVASCOLARI
– RIDOTTA ELIMINAZIONE RENALE
• SOLUBILITA' AUMENTATA (DICUMAROLI, CLOFIBRATO)
• DURATA D'AZIONE MAGGIORE
– DIGITOSSINA (t/2 7gg) vs K STROFANTINA (t/2 12 h)
• POSSIBILITA’ DI SPIAZZAMENTO
– AUMENTA LA QUOTA DI FARMACO LIBERO (IMPORTANTE PER
FARMACI FORTEMENTE LEGATI, CON BASSO INDICE TERAPEUTICO E
CON BASSO VOLUME DI DISTRIBUZIONE es antiaritmici, anticoagulanti
tipo warfarin)TIPI DI LEGAME: •elettrostatici •Van der Waals •idrofobici •idrogeno
4. AFFINITA' TISSUTALE
• ACCUMULO tissutale per effetto di legame, in genere reversibile, ai
costituenti tissutali (proteine, prot nucleari, fosfolipidi).
• Effetto: riserva di farmaco nel tessuto di accumulo, o in un sito lontano,
raggiunto dalla circolazione
• Può anche essere causa di tossicità (es aminoglicosidi e ototox)
• Accumulo nel tess adiposo di ff liposolubili (es tiopentale, remifentanile,
alcuni beta-bloccanti)
• Accumulo nell’osso:
– tetracicline e metalli pesanti (tox)
– Fosfonati e accumulo nell’osso (eff terapeutico)
5. PERMEABILITA'
• La permeabilità degli endoteli varia a seconda del distretto considerato
(max sinusoidi epatici, min BEE)
6. BARRIERE ANATOMO FUNZIONALI
• barriera emato-encefalica
• barriera placentareAREE CEREBRALI PRIVE DI BEE • epifisi • eminenza mediana • CTZ • area postrema del IV ventricolo
Passaggio dei ff nel SNC
• Il passaggio dei ff nel SNC è determinato dalla liposolubilità del
composto, dal suo grado di ionizzazione, e dalla presenza di
trasportatori.
• Ostacolato dalla presenza di giunzioni serrate negli endoteli
cerebrali (BEE) e di trasportatori di efflusso, che estrudono il
farmaco:
– glicoproteina-P codificata dal gene MDR1
– Polipeptide trasportatore degli anioni organici (OATP)
• Può essere facilitato da particolari formulazioni (es liposomi o solid-
liquid nanoparticles)
• L’integrità della BEE viene alterata da infiammazioni e meningiti (
facilita il passaggio)
• La compartimentalizzazione può essere sfruttata terapeuticamente:
– Antistaminici II (non sedativi)
– LoperamideVOLUME APPARENTE DI DISTRIBUZIONE (Vd)
• Volume teorico di acqua corporea richiesto per contenere la quantità di farmaco presente nell’organismo,
supponendo che la sua concentrazione sia ovunque uniforme e pari alla concentrazione plasmatica.
• Espresso sia come Volume totale (in litri) che come volume/Kg peso corporeo
• COME SI DETERMINA
• Iniezione di quantità nota di sostanza. Prelievo di sangue (dopo un certo periodo di tempo)
• Determinaz della concentraz plasmatica (o nel sangue in toto)
• Conc (mg/l) = Dose (mg)/ Vol (l) Vol = Dose/ conc
• Volumi corporei
– Plasma(3 litri)
– Sangue (5.5 liri)
– Liquidi extracell (escluso plasma) 12 litri
– Acqua corporea 42 litri
• Quindi:
• Vd = 3 l sost ad elevato PM che si distribuiscono nell’acqua plasmatica (es BLU EVANS)
• Vd = 42 l sost a diffusione omogenea, (es D2O)
• Vd = 13 l Sost che si distribuiscono nei liquidi extracellulari (es INULINA)
• Sostanze molto liposolubili o con elevata affinità tissutale hanno Vd elevatissimi
• Digossina 210 l, Diazepam 77 l
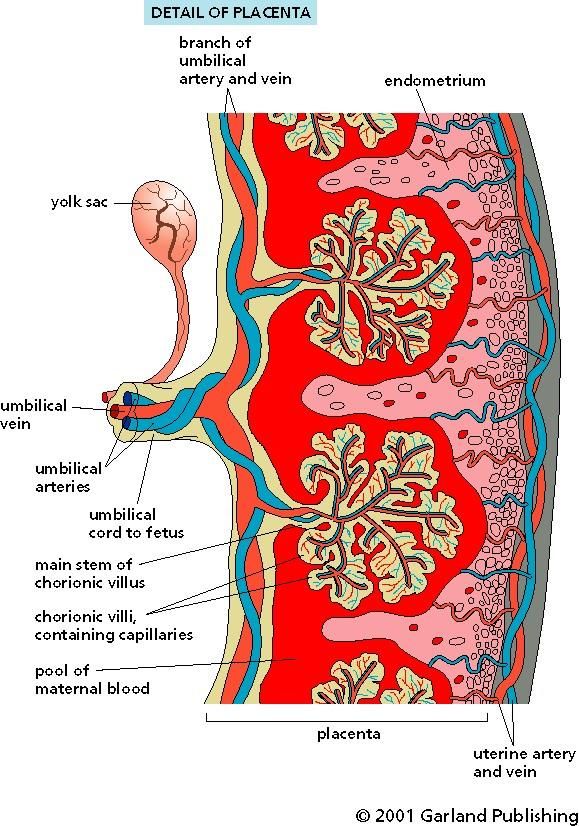
• I farmaci molto legati alle proteine plasmatiche hanno Vd bassi (purchè non siano anche distribuiti ai tessuti)PLACENTA
ANNESSO EMBRIONARIO DEPUTATO ALLA
NUTRIZIONE ED OSSIGENAZIONE DEL
FETO ED ALLA ELIMINAZIONE DEI SUOI
PRODOTTI DEL CATABOLISMO
NON E’ UNA VERA BARRIERA
Meccanismi di passaggio:
Il sangue proveniente dal feto raggiunge la
DIFFUSIONE PASSIVA placenta per mezzo delle arterie ombelicali,
(molecole idro-liposolubili) scorre nei villi e ritorna al feto attraverso la vene
TRASPORTO ATTIVO O FACILITATO ombelicale.
(aminoacidi, I-, vitamine) Il sangue materno entra nello spazio intervilloso
PINOCITOSI (immunoglobuline) attraverso le arterie spiraliformi e ritorna nel
circolo attraverso molte vene. Il sangue fetale e
quello materno non si mescolano.FATTORI CHE INFLUENZANO IL PASSAGGIO
TRANSPLACENTARE
• liposolubilità e ionizzazione (pH del plasma fetale inferiore
a quello materno di 0.1-0.2 unità)
• peso molecolare
< 500 passaggio agevole
> 1000 passaggio impossibile
• legame alle proteine plasmatiche materne
• flusso sanguigno utero-placentare
fumo! riduce la perfusione placentare
vasocostrizione da nicotina o cocainaMetabolismo
Insieme di reazioni enzimatiche finalizzate alla
biotrasformazione delle sostanze esogene (xenobiotici) in
composti più polari ed idrosolubili, più facilmente
eliminabili dall’organismo.
Infatti
Le caratteristiche chimico-fisiche che favoriscono
l’assorbimento di un farmaco o di un tossico sono in genere
diverse da quelle che ne favoriscono l’eliminazione.
…NON SOLO FARMACI….Xenobiotico
Bioattivazione
detossificazione Bioattivazione
(profarmaco)
Metaboliti stabili Metaboliti attivi Metaboliti reattivi
Effetto terapeutico
Tossicità
Es epatotox da Parac
Cancerog da benzene
teratogenicitàda un composto attivo si può formare
un composto inattivo (detossificazione)
composto attivo con diversa durata d’azione
composto tossico stabile o intermedi reattivi
(bioattivazione)
da un composto inattivo si può formare
un composto con attività farmacologica o
un composto con attività tossica
(bioattivazione)perché è importante studiare il metabolismo
in farmacologia/ tossicologia
stabilire e adattare la posologia
evidenziare la presenza di altri composti (attivi, inattivi, tossici) e prevedere
nuove sintesi
prevedere interferenze metaboliche con altri farmaci
prevedere variazioni di risposta in seguito a trattamento protratto
(induzione e repressione enzimatica)
evidenziare i meccanismi di tossicità degli xenobiotici
prevedere riposte idiosincrasiche in base al profilo enzimatico individuale
prevedere la suscettibilità individuale a xenobiotici presenti nell’ambiente, nei
luoghi di lavoro etc..localizzazione delle reazioni metaboliche
metabolismo presistemico (prima dell’assorbimento)
prevalenti reazioni di idrolisi e riduzione
ma anche b-glicuronazione e nitrosazione
eliminazione presistemica (o first pass epatico)
metabolismo sistemico
extracellulare
(enzimi plasmatici; esterasi e amidasi)
intracellulare
enzimi
citoplasmatici
mitocondriali
microsomialiFASI
FASE I
(funzionalizzazione; inserzione o messa in evidenza di gruppi funzionali)
ossidazioni
(perossidazioni)
riduzioni
idrolisi
idratazioni
detioacetilazioni
isomerizzazioni
FASE II
(coniugazione o sintesi; coniugazione con sostanze per rendere la molecola più idrofila)
acetilazioni (N-acetil transferasi, NAT)
coniugazioni con
ac glicuronico (UDP-glucuronil transferasi, UGT)
glutatione (Glutatione-S-transferasi, GST)
glicina ed altri aa
solfatazioni (solfotransferasi, SULT)
metilazioni (metiltransferasi, MT)
condensazioni
FASE III
A carico dei coniugati con glutatione, escreti per via urinaria dopo metabolizzazione ad acidi
mercapturiciOssidazioni
Responsabili della metabolizzazione di circa l’80 % dei farmaci
Microsomiali e mitocondriali
Monossigenasi citocromo P450 dipendenti
Mediano C- e O-ossidazione, dealchilazione, etc
Monossigenasi flavina dipendenti (FMO)
Mediano N-, S-, e P-ossidazione
Epossido idrolasi
Mediano idrolisi epossidi
Deidrogenasi (citosoliche)
Alcoli e aldeidiCLASSIFICAZIONE E NOMENCLATURA DEI CITOCROMI 300 geni, circa 1000 enzimi, con isoforme Nell’uomo 57 geni e 58 pseudogeni analogia di struttura < 40 % famiglie es CYP1 40-55 % sottofamiglie es CYP1A >55 % sottotipi es CYP1A1 Localizzazione Fegato Epitelio esofago, stomaco, intestino Cellule Clara dei polmoni Cellule tubuli prox rene gh surrenali, epitelio vescica SNC (cute, placenta e linfociti) NO attività P-450 dip: m scheletrico, cuore, utero, milza ed eritrociti
Ossidazioni a funzione mista (microsomiali, mitocondriali) (dalla mol di O2 attivata un atomo viene ridotto a acqua ed un altro viene donato al substrato) Viene consumato O2 in eccesso con formazione di O-2 poi detox dalle SOD
specificità • i CYP che metabolizzano gli xenobiotici hanno scarsa selettività di substrato, c’è spesso sovrapposizione, e bassa velocità di catalisi. • Base per interaz tra farmaci. • I CYP sono coinvolti nella sintesi di ormoni (es CYP19 o aromatasi, converte il testosterone in estrogeni), dei Sali biliari, degli eicosanoidi, dell’ac retinoico. I CYP che sintetizzano composti endogeni hanno elevata specificità di substrato
12 famiglie di CYP nell’uomo: i principali sono
enzima Organo Substrato Frazione inducibilità
relativa al
CYP epatico
CYP1A1 Polmone, IPA 0
placenta, pelle
CYP1A2 Fegato Amine aromatiche 5 50 volte
Caffeina
Aflatossina B1
CYP2A6 Fegato Curarina• Metabolismo dei farmaci • CYP3A4/5 (50 %) • CYP2D6 (15 %) • CYP2C8/9 (10 %) • I CYP1A1/2, CYP1A6, CYP1B1/2B1, CYP2E1 non sono coinvolti in metabolismo di farmaci ma bioattivano una serie di xenobiotici. • E’ plausibile che regolino la suscettibilità individuale ai cancerogeni • CYP di altre famiglie sono coinvolti nel metab delle sostanze endogene (steroidi, ac grassi, vitamine, etc)
Metabolismo del 60% 30% paracetamolo 5% Le vie di detossificazione sono ad alta affinità ma a bassa capacità: NABQI saturazione!
• Reazioni di fase II • (In genere detossificanti. Eccezioni: morfina M6G) • Rapida velocità di catalisi • CONIUGAZIONE CON AC GLICURONICO • UDP-glicuronil transferasi (cof ac UDP glicuronico) • MICROSOMIALE (fegato, intestino, rene, cute, cervello, polmone, in stretta associazione col sistema CYP) • Inducibile, presenza di isoforme • I glicuronoconiugati sono escreti per via renale e biliare in base al PM dell’aglicone (se 350 eliminaz biliare) • La scissione di glicuronidi nell’intestino dovuta alle beta-glicuronidasi batteriche può prolungare la permanenza degli xenobiotici nell’organismo (circolo entero epatico). • UGT1A1 metabolizza la bilirubina. S. di Gilbert (effetti tossici dopo assunzione di ff) è associata a ridotta espressione di UGT1A1 per mutazione nel promotore: • Elevata bilirubinemia • Tox da irinotectan
• CONIUGAZIONE CON GLUTATIONE • Unico caso di reaz con composti elettrofili • GSH S- transferasi (cof GSH; cys-glu-gly) • isoforme • MICROSOMIALE e CITOPLASMATICA (diversi substrati) • I glutatione-coniugati sono escreti per via biliare o trasformati ad ac mercapturici (raz FASE III) • Il GSH è contenuto in tutte le cellule (fino a 10 nM) • Altri ruoli del GSH: • Scavenger • Trasporto di composti endogeni • Mantenimento dei sistemi redox
acetilazioni • N-acetiltransferasi (NAT) citosoliche (amine aromatiche, idrazine) • Cofattore acetil CoA • Fenotipo acetilatore: INI ed effetti tossici (5-15%) • Lento e rapido acetilanti. Nascita della farmacogenetica • NAT1 e NAT2 (polimorfismi di NAT2 sono responsabili del fenotipo lento acetilante) • I lento acetilanti possono sviluppare malattie autommuni da farmaci (es LES) e vanno incontro a sintomi di sovradosaggio dopo somm di alcuni farmaci: • Idralazina, INI, sulfamidici, amantadina, etc
FATTORI CHE MODIFICANO IL METABOLISMO
1. POLIMORFISMO GENETICO
2. INTERAZIONE CON ALTRI XENOBIOTICI
3. PATOLOGIE
4. ETA’ e GENERE
5. ABITUDINIFATTORI CHE MODIFICANO IL METABOLISMO
• 1. POLIMORFISMO GENETICO
• Individui con diverse capacità metaboliche per la presenza di isoenzimi
con attività ridotta o aumentata.
• Conseguenze:
• Variate risposte ai farmaci su base quantitativa e qualitativa
• (risp idiosincrasiche)
• Tossicità ed effetti terapeutici
• Suscettibilità a cancerogeni
• Polimorfismi di interesse clinico:
– NAT1 e 2
– Alcol e aldeide deH
– Locus Ah
– CYP2D6, CYP2C9, CYP2C19Rilevanza farmaco-tossicologica dei polimorfismi genetici
• Polimorfismo genetico: presenza di due o più varianti alleliche (e fenotipiche) con frequenza >1 % in una
popolazione di individui.
• 1960. Prima evidenza che il polimorfismo degli enzimi avesse rilevanza clinica (lento acetilanti e
isoniazide)
• 1977. prima evidenza di polimorfismo dei citocromi (debrisochina e CYP2D6, “lento idrossilanti”)
• Si possono differenziare individui con:
• A. 2 alleli che codificano per l’enzima “normale”
(metabolizzatori estensivi omozigoti; homozygous extensive metabolizers, EM, wild-type)
• B. 1 allele normale e 1 allele che codifica per un enzima a funzione ridotta
(metabolizzatori estensivi eterozigoti; metabolizzatori intermedi; intermediate metabolizers, IM)
• C. 2 alleli che codificano per un enzima con attività ridotta o nulla
(lento metabolizzanti, poor metabolizers, PM)
• D. Duplicazioni o moltiplicazioni geniche (per ora identificate solo per CYP2D6)
(ultrarapid metabolizers; UM)• L’impatto clinico dei polimorfismi dipende da alcuni fattori:
• contributo della specifica isoforma al metabolismo del farmaco (es presenza di vie di
metabolizzazione alternative)
• presenza di metaboliti attivi
• presenza di enantiomeri (spesso gli enantiomeri hanno vie metaboliche differenti)
• conversione fenotipica (phenocopying)
(es un pz EM può trasformarsi in PM per auto- o etero-inibizione enzimatica
vd interazione tra farmaci
Qs conversione può avvenire durante la terapia e quindi l’impatto della farmacogenetica
può variare dalle condizioni iniziali rispetto a quelle di mantenimento.
• dose del farmaco (es aloperidolo è metabolizzato a basse dosi dal CYP2D6 e a dosi elevate
da CYP3A4)
• Quindi in molti casi l’impatto clinico derivante dalla presenza di polimorfismi
genetici è clinicamente irrilevante.
• Le condizioni possono cambiare quando:
• 1. gli effetti del polimorfismo sui siti attivi dell’enzima è drammatico (es PM)
• 2. Si dimostra una chiara associazione tra concentrazione ed effetto terapeutico
• 3. Si dimostrano chiari e severi effetti avversi concentrazione-dipendenti
• 4. Il farmaco ha basso IT• Principali polimorfismi a carico dei citocromi
• CY2D6
• CYP2C9
• CY2C19CYP2D6
• Coinvolto nell’eliminazione di ~25% dei farmaci
(alcuni beta-bloccanti, antidepressivi, neurolettici, antiaritmici, oppioidi)
• Il più studiato in relazione ai polimorfismi
• 7-10 % dei caucasici sono PM
• Principali alleli inattivi *3, *4, *5
• Nei caucasici la principale variante è la *4 (~21%)
• Nei cinesi la principale variante è la *10 (~50%) che genera enzimi ad attività ridotta (non
assente)
• Duplicazione/moltiplicazione genica osservata in ~1 %degli svedesi, ~7% degli spagnoli e
~29% etiopi neri
• Casi clinici rilevanti
• Dove appare utile la genotipizzazione per predire effetti avversi o risposte cliniche
• 1. In un lavoro (Rau 2004) è stato osservato che il 29 % dei pz con effetti avversi da TCA
erano PM mentre il 19 % dei non responders erano UM
• 2. Codeina. La trasformazione in morfina è dovuta al CYP2D6. scarso effetto analgesico in
PM. Esagerata risposta in UM (abuso?)
• 3. atomoxetina (STRATTERA®). Inibitore reuptake NE usato in ADHD.
Nel foglietto illustrativo è riportato che nei PM possono essere sperimentati effetti avversi
con frequenza significativamente (2-3 volte) maggiore che negli EMCYP2C9
• E’ il più abbondante nella famiglia CYP2C
• Rappresenta circa 33% dei CYP epatici.
• Coinvolto nel metabolismo di >100 farmaci tra cui anticoagulanti dicumarolici,
sulfoniluree, inibitori angiotensina II, alcuni FANS.
• *2, *3 sono le varianti alleliche che generano enzimi a ridotta attività più
frequenti (almeno un allele in 20 % e 12 % caucasici, rispett, 2.5 % omozigoti per
una delle due)
• Casi clinici rilevanti
• 1. Warfarina (enantiomero S)
• I pz con *2 e *3 richiedono meno farmaco.
• Stretta associazione con varianti alleliche del gene che codifica per la proteina
target dei dicumaroli: la vitamina K epossido reduttasi (VKORC1).
• Polimorfismi di CYP2C9 e VKORC1 assieme a variabili quali l’età ed il peso
possono giustificare il 50-60 % delle richieste posologiche.
• 2. Fenitoina
• PM hanno livelli più elevati: tossicità!CYP2C19 • PM in 1-8 % caucasici • Principale variante con prodotto inattivo è la *2 (75% dei PM) • Farmaci metabolizzati: inibitori di pompa (omeprazolo, lansoprazolo e pantoprazolo), BZD, TCA, SSRI, fenitoina. • PM hanno AUC 4-16 volte più elevate per omeprazolo. No effetti avversi a causa dell’alto IT. Anzi migliore soppressione acida e migliore eradicazione di H pilori.
• 2. INTERAZIONE CON ALTRI XENOBIOTICI Repressione e induzione enzimatica • Il metabolismo degli farmaci/tossici può essere variato dall’esposizione a farmaci, micronutrienti, sostanze presenti nell’ambiente • L’interazione può avere importanti ricadute cliniche di natura farmaco/tossicologica • Inibizione farmacometabolica • Induzione farmacometabolica
• INIBIZIONE FARMACOMETABOLICA
• Processo per cui l’esposizione a due sostanze porta all’inibiz del metabolismo di un farmaco
e all’aumento della sua conc con possibili effetti tossici
• Pericoloso per sostanze rapidamente metabolizzate e con basso IT
• Insorgenza rapida
• Meccanismi
• Competizione per lo stesso enzima (la sostanza che interferisce può essere substrato o
meno)
• Inattivazione dell’enzima (inibitori suicidi)
Es CCl4
• Legame all’eme
Ketoconazolo
• Sostanze che riducono la sintesi di cofattori
Es reaz fase II
• Es clinicamente rilevanti: Inibitori CYP3A
Antifungini azolici (interferenza con terfenadina!!!)
Inib proteasi HIV
Antibiotici macrolidi
Succo di pompelmo
Ca antagonistiINDUZIONE FARMACOMETABOLICA • preval a livello epatico, • a carico di CYP ma anche di altri enzimi microsomiali (es UGT) • insorgenza lenta • meccanismi: • induzione enzimatica • stabilizzazione della proteina alla degradazione (CYP2E1) • conseguenze • tolleranza farmacometabolica (autoinduzione, es carbamazepina) • riduz concentrazioni attive di farmaci metabolizzati dallo stesso CYP (eteroinduzione) con perdita dell’efficacia terapeutica (es rifampicina + contacc orali) • aumentata velocità di bioattivazione (es paracetamolo + alcol epatotox)
• 3. PATOLOGIE ES epatopatie • 4. ETA’ e GENERE La maturazione funzionale degli enzimi metabolici si completa entro la seconda decade di vita e declina con l’età. Risposte diverse tra i sessi (di scarso rilevo clinico) Eccezione: gravidanza (durante il 2 e 3 trimestre alcuni enzimi vengono indotti così da richiedere aggiustamenti posologici) • 5. ABITUDINI Fumo, caffè, dieta
Eliminazione dei farmaci • Farmaci escreti in forma immodificata, o metabolizzati
• Principali emuntori:
Rene, fegato, polmone (anestetici gassosi)
• Altre vie di eliminazione: saliva, sudore,
lacrime, cute e capelli, latte materno
• gli emuntori eliminano più facilmente ff con
elevata polarità. I ff liposolubili devono quindi
essere trasformati (eccez polmone)• CLEARANCE D'ORGANO
• QUANTITA' DI SOSTANZA RIPULITA DA QUELL'ORGANO NELL'UNITA' DI
TEMPO
• CL= FLUSSO x INDICE D'ESTRAZIONE (in ml/min)
• INDICE D'ESTRAZIONE = Cart - Cven
Cart
FLUSSO PLASMATICO RENALE 650 ml/min
FLUSSO PLASMATICO EPATICO 800ml/min
• la clearance sistemica totale di un farmaco è la somma delle diverse CL nei
vari organi nei quali il farmaco è eliminato.
• corrisponde alla sua velocità di eliminazione, attraverso tutte le vie,
normalizzata per la conc di farmaco
– CL= velocità di eliminazione/Conc plasmatica farmaco• Per la maggior parte dei farmaci, la CL è una funzione
lineare della concentrazione plasmatica (in quanto gli
enzimi metabolizzanti o i trasportatori non sono
saturati). Cinetica di primo ordine, cioè una frazione
costante di farmaco è eliminata nell’unità di tempo
– All’equilibrio la CL di un farmaco (con biodisponibilità
completa e cinetica di primo ordine) è determinata dal
rapporto CL=Dose/AUC
• Nei casi in cui i sistemi di metabolismo/trasporto
vengano saturati, l’eliminazione segue una cinetica di
ordine zero, secondo cui una QUANTITA’ costante di
farmaco è eliminata nell’unità di tempoELIMINAZIONE RENALE
• La funzione renale è bassa nei neonati e negli anziani
La velocità e l’entità dell’eliminazione renale dipendono da:
• 1. Filtrazione glomerulare (proporzionale alla permeabilità
dei capillari e pressione sanguigna)
– Velocità di filtrazione glomerulare 125 ml/min
– Fattore limitante è il PM (60.000) i tossici legati alle proteine
non sono filtrati
• 2. Riassorbimento tubulare ATTIVO (es glucosio) e PASSIVO
(pH dipend, sostanze liposolubili, secondo gradiente conc)
• 3. Secrezione tubulare via glicop-P, o proteina MRP2 (es PAI)
• Clearance compresa tra 0 (ultrafiltrato e riassorbito
completam) e 650 (ultrafiltrato+secreto).
• Inulina = 125 (solo ultrafiltrato)
• Eliminazione pH dipendente per acidi e basi deboliMisura della funzionalità renale
• La creatina è un prodotto di degradazione della fosfocreatina muscolare.
• E’ eliminata esclusivamente per via renale
• Clearance della creatinina
Creatinina urinaria x Volume urinario (24 ore) / Creatinina serica
Valori nomali 85-125 ml/min
• Anche la semplice misura della creatininemia dà un indice di funzionalità
renale
• VALORI NORMALI UOMO: 0,8-1,4 mg/dl (Europa e SI: 70-123 umol/L)
• VALORI NORMALI DONNA: 0,6-1,2 mg/dl (Europa e SI : 53-106 umol/L)
• VALORI NORMALI BAMBINO: 0,2-1,0 mg/dl (Europa e SI : 18-88 umol/L)ESCREZIONE fecale e biliare
• I ff che ritroviamo nelle feci sono:
– Farmaci dati per os e nn assorbiti
– Metaboliti secreti direttamente nelle feci
– Metaboliti escreti per via biliare
• Produzione biliare 0.5-1 litri 24 ore
• Meccanismo: ultrafiltrazione, secrezione (riassorbimento)
• Composizione: ioni e glucosio (conc = plasma), proteine e lipidi
• Gli epatociti possiedono molti trasportatori:
– P-gp e BCRP (ABCG2) trasportano molti ff liposolubili
– MRP2 metaboliti coniugati
• L'ELIMIN BILIARE E' GOVERNATA DA
– POLARITA' (direttam prop)
– PM (secreti solo composti con PM 300-500 e glicurono-coniugati)
– FENOMENI DI TRASPORTO ATTIVO
•
• NELLA BILE troviamo SOSTANZE LA CUI CONC RISPETTO AL PLASMA E‘
– UGUALE (es, glucosio, Na+, K+, Hg++, Co++) CLASSE A
– MAGGIORE (10-100 volte) (es. ac. biliari, bilirubina, solfobromoftaleina, Pb++, Mn++) CLASSE B
– MINORE (es. albumina, inulina, Zn++, Fe++, Cr++) CLASSE CCircolo entero-epatico • I ff possono essere eliminati con la bile ma riassorbiti (anche con gli stessi trasportatori) • Il circolo ee prolunga l’azione dei farmaci • Il circolo ee prolunga l’azione delle tossine (es resina a scambio ionico e intox Hg o glicosidi)
emivita
Emivita è il tempo necessario affinchè:
• la quantità totale di farmaco nell’organismo si dimezzi (e. biologica)
• La concentrazione plasmatica del farmaco nel plasma si dimezzi (e.
plasmatica, T1/2, plasma half-life, detta anche emivita terminale o emivita di
eliminazione). Si calcola in condizioni di equilibrio, quindi quando dipende solo
dall’eliminazione
• E’ inversamente proporzionale alla costante di eliminazione (k), cioè della
frazione di farmaco eliminata nell’unità di tempo.
• Es k=0.1 * h-1 (=un decimo del farmaco eliminato in 1 h) (k=CL/Vd)
• T1/2 =0.7 Vd/CL
• L’emivita determina:
– Durata d’azione dopo singola somm (empiricamente il raddoppio della dose
provoca un aumento della durata d’azione pari a una emivita)
– Tempo richiesto per raggiungere lo stato-stazionario o steady state durante terapia
cronica per infusione a vel costante (empiricam servono 3-5 emivite per
raggiungere lo steady state)
– Frequenza di sommPuoi anche leggere

















































