Siti Fragili del cariotipo umano - eLearning
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Siti Fragili del cariotipo umano
1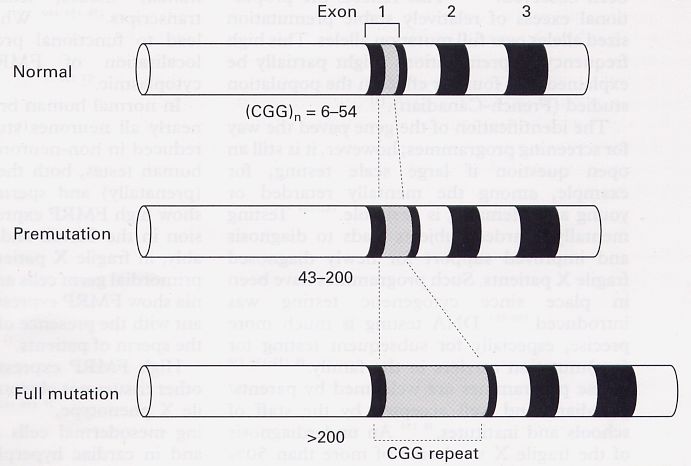
Oggi I siti fragili sono definiti come…
… punti sui cromosomi che presentano interruzioni o rotture non casuali quando le cellule sono
esposte a determinati agenti chimici o a specifiche condizioni di coltura.
… regioni cromosomiche il cui DNA non ha compiuto il normale compattamento mitotico oppure
ha subito una despiralizzazione prematura (Sutherland, 1991).
Allestimento di metafasi da linfociti coltivati per errore in terreno TC199 un terreno povero in acido folico e timidina.
Presenza di rotture in siti non casuali.
Il termine “sito fragile” viene per la prima volta usato da
Hecht* in riferimento ad una rottura ricorrente sul braccio
lungo del cromosoma 16 (16q)
2
* Magenis, Hecht, Lovri En : Heritable fragile site on chromosome 16: probable localization of haptoglobin locus in man.
Science 170:85-87, 1970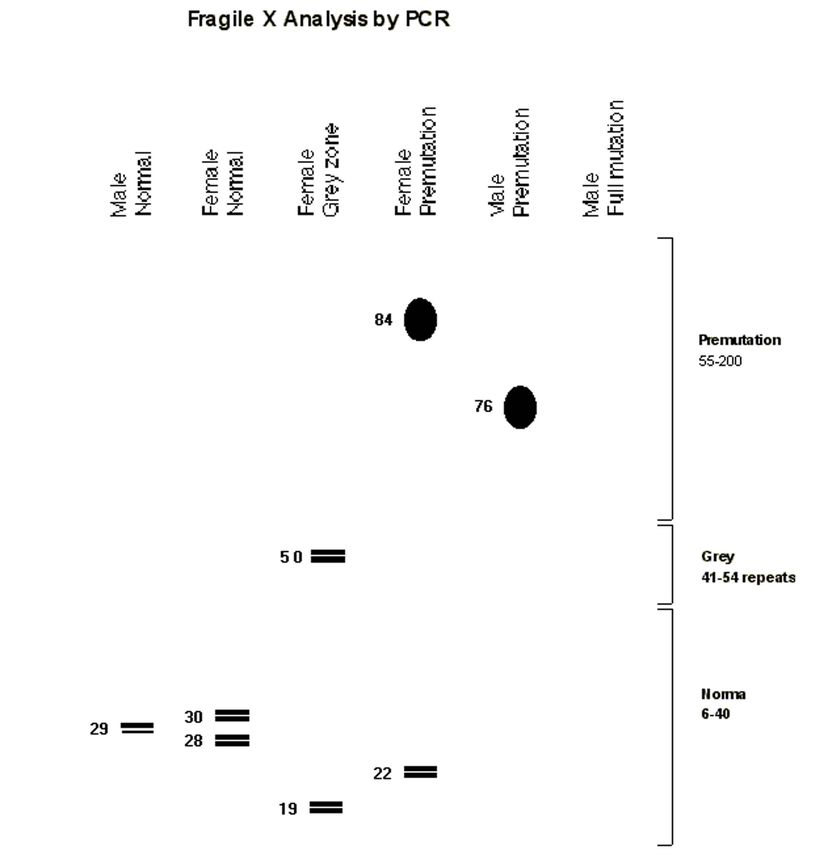
Sutherland et al. 1985
Negli stessi anni all’interno di piccole famiglie un sito
di fragilità viene associato ad una forma di ritardo
mentale.
In queste famiglie il sito fragile nella regione Xq28
(Xq27.3) viene ereditato in associazione con il
ritardo mentale.
Il sito fragile viene osservato in metafasi di individui
affetti dalla patologia coltivando le cellule in
particolari condizioni: in assenza di acido folico nel
terreno di coltura.
Successivamente si comprese che lo stress indotto
dall’assenza di acido folico ricadeva sul
metabolismo delle basi puriniche e pirimidiniche,
inibendo parzialmente la replicazione del DNA,
L’aggiunta di acido folico al terreno TC199
causando le rotture e non condensazioni visibili sui
riduce sensibilmente le rotture nelle regioni cromosomi metafasici.
fragili
3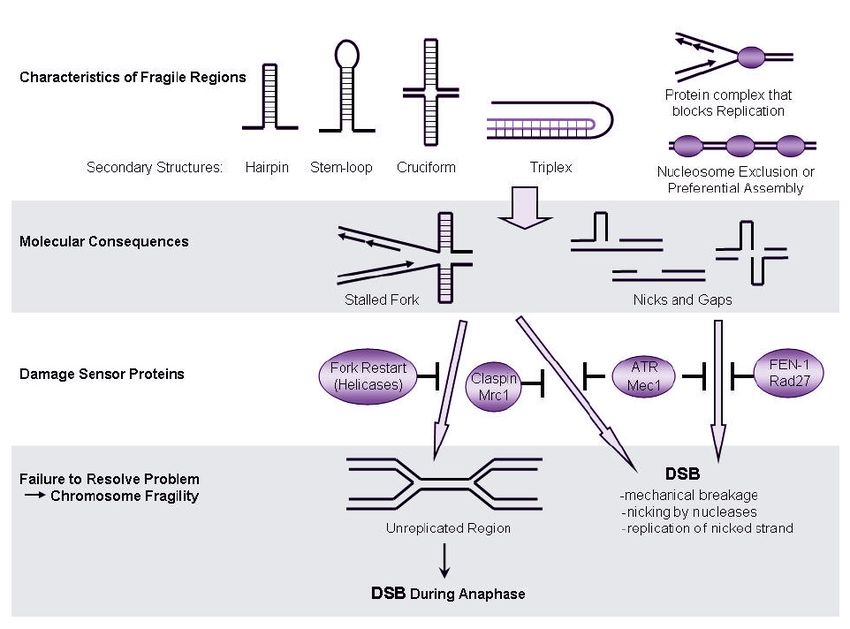
Metabolismo cellulare
dell’acido folico
timidina
monofosfato
folato - cofattore per la conversione
dell’uridina monofosfato in timidina
FRAXA
(UMP)
uridina
monofosfato 4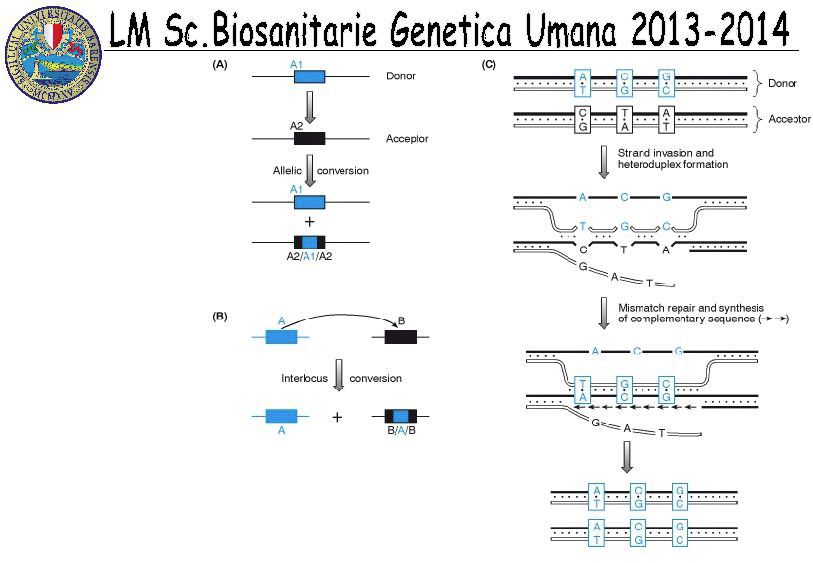
Queste discontinuità possono presentarsi unite da un sottile filamento di materiale visibile
al microscopio elettronico; in alcune metafasi invece si verifica una vera e propria rottura
in corrispondenza del sito fragile.
Sebbene i siti fragili siano talvolta espressi spontaneamente nei preparati cromosomici da
colture di linfociti, quasi tutti richiedono un’alterazione delle condizioni di crescita in
grado di permetterne o di aumentarne l’espressione (Sutherland, 1991).
Nomenclatura
es. FRA2G
2q24.3-31.1
5
Siti fragili
RARI o ereditabili COMUNI o costitutivi
Classificati in base alla frequenza con cui si ritrovano nella popolazione e in base alla loro ereditarietà
Siti fragili rari
- sono presenti in una piccola percentuale della popolazione (< 5%);
- sono ereditati come carattere mendeliano semplice.
Distamycin
6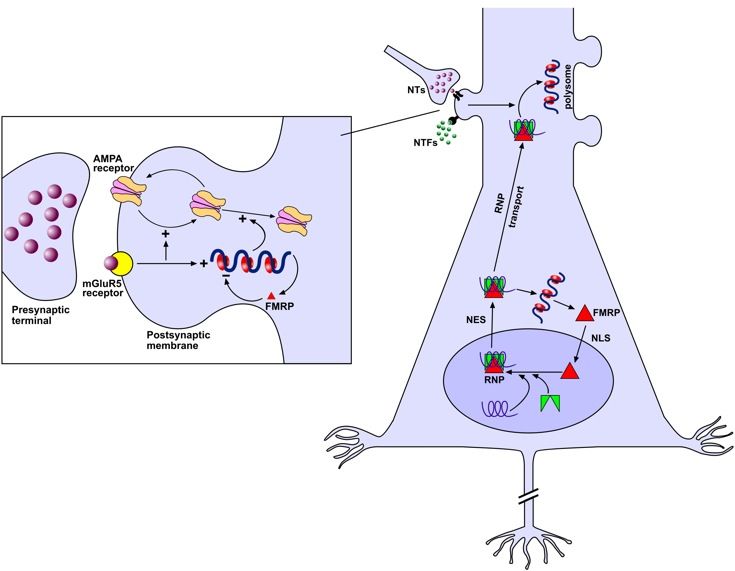
I siti fragili rari folato sensibili
Tutti i siti fragili rari indotti dal folato fino ad oggi caratterizzati presentano al loro interno una
ripetizione di trinucleotidi CGG il cui numero può variare nei vari individui.
Il numero delle ripetizioni è responsabile dell’espressione del sito fragile.
⇓
L’espressione dei siti fragili rari è stata associata ad una particolare classe di mutazioni, definite
mutazioni dinamiche, in cui una ripetizione di trinucleotidi può essere soggetta ad espansione a causa di
diversi meccanismi molecolari nel corso delle generazioni
SINDROME DI MARTIN-BELL
“SINDROME DELL’ X FRAGILE”
• forma più comune di ritardo mentale dopo la sindrome di Down e la più frequente fra quelle ereditarie
Sintomi
• ritardo cognitivo ( incapacità linguaggio - ritardo mentale)
• deficit d’attenzione e iperattività
• ansia e umore instabile
• ritardo nello sviluppo psicomotorio
Tratti somatici
• viso stretto e allungato
• fronte e mandibola prominenti
• orecchie grandi e piedi piatti
• macrorchidismo
➛ Coltivando le cellule di individui affetti da sindrome dell’X-fragile in un terreno povero di acido
folico, si osserva in una percentuale variabile di metafasi, l’espressione di FRAXA
Causa
• è causata dall’alterazione del gene FMR1 (Fragile Mental Retardation 1) situato sul cromosoma
• (q27.3)Ipermetilazione della regione 5’-UTR del gene (CpG)
FMR1p
Lubs, A marker X chromosome. Am J Hum Genet.
1969 May; 21(3): 231–244.
FIG. 1.-Pedigree showing that each of the four retarded males had the marker X chromosome. Two females with the
chromosome were normal. A third (II-3) must have transmitted the chromosome to her son, but the secondary
constriction was not observed in 250 cells.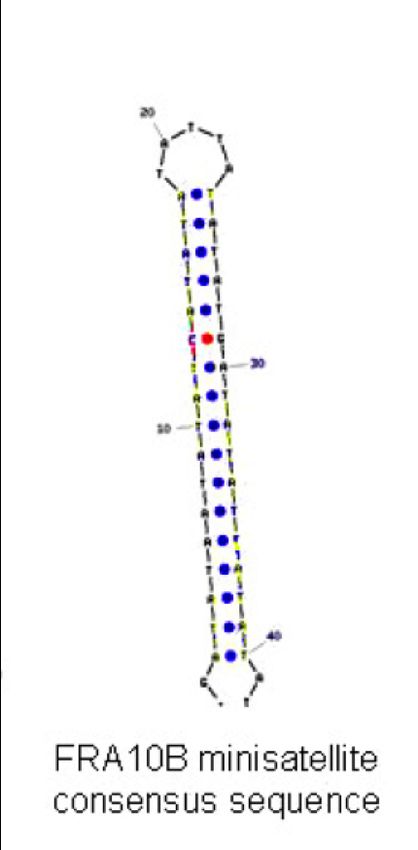
Lubs, A marker X chromosome. Am J Hum Genet. 1969 May; 21(3): 231–244. FIG. 3.-Karyotype from the proband showing the FIG. 4. The “satellites” appeared symmetrically marked secondary constriction present in at the end of the chromatids (left), with one-third of his cells. displacement of one satellite to the other The resulting satellites are smaller in diameter chromatid (left center), as a single centrally than the chromatid but larger than any of the placed satellite (center) or rarely as tandem satellites on the acrocentric chromosomes. satellites (right).
Lubs, A marker X chromosome. Am J Hum Genet. 1969 May; 21(3): 231–244. H3-thymidine was added to one bottle from each culture four hours prior to harvesting Fig. 5 Autoradiograph from III-I. The Karyotype with the emulsion in place is shown below the usual karyotype. The only chromosome with heavy labeling also was the chromosome with satellites (lower right).
The FMR1 Gene
Fragile X syndrome (FXS) is the most common
known cause of inherited intellectual disability.
FXS affects both males and females. However,
females often have milder symptoms than males.
Number of people who have FXS is unknown, but
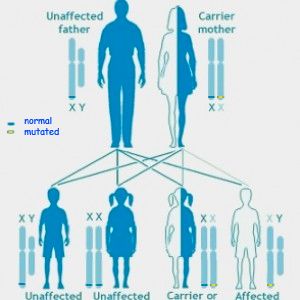
it has been estimated that about 1 in 5,000 males Fragile X is Inherited
are born with the disorder.
Each child of a carrier woman has a 50% chance of
inheriting the mutated gene.
When the stretch of DNA expands beyond a A mother who is a carrier of Fragile X has a 50%
certain length, the gene is switched off and does chance of passing the mutated gene on to each of her
not produce the protein that it is normally makes. children. Those children can in turn also be carriers or
This gene change is called a “full mutation”. A male they might be fully affected with Fragile X syndrome.
who inherits a full mutation exhibits Fragile X Carrier men will pass the premutation to all their
syndrome because his only X chromosome contains daughters but none of their sons. These daughters are
the mutated gene. A female may not be as severely carriers but they do not have Fragile X syndrome. The
affected because each cell of her body needs to Fragile X premutation can be passed silently down
through generations in a family before a child is born
use only one of its two X chromosomes and
with the syndrome.
randomly inactivates the other.The FMR1 Gene
The most common fragile X mutation is an expansion
of extra DNA (CGG)n within a part of the FMR1
gene. In the premutation, the expansion is relatively
small. In the full mutation the expansion is quite
large and is usually accompanied by abnormal
methylation, which leads to decreased production or
absence of the FMR1 gene’s protein product, called
FMRP.
FMR1
• 17 esoni
• lungo 38 Kb
• tripletta CCG ripetuta nel 1° esone
• il numero di ripetizioni della tripletta CCG è polimorfico tra gli individui normali
• il numero di ripetizioni della tripletta CCG del 1° esone determina lo stato di metilazione del gene
12FMR1 e il ritardo mentale
• FMR1p ha una funzione neurone specifica: è in grado di legare specifici RNA riconoscendo una
sequenza consenso di legame
• Regola la localizzazione e la traduzione di specifici mRNA essenziali nelle sinapsi nervose
• L’assenza di FMR1p causa una diminuizione dello stimolo sinaptico
• La perdita della funzione sinaptica ottimale potrebbe spiegare il ritardo mentale presente negli
individui che esprimono FRAXA
Oostra & Willemsen, A fragile balance: FMR1 expression levels. Hum Molec Genetics. 12(suppl. 2):R49-R57, 2003. doi: 10.1093/hmg/ddg298
Modello molecolare
NTs: neurotransmitters
NTFs: neurotrophic factors
NLS: nuclear localization signal
NES: nuclear export sequence
AMPA: (α-amino-3-hydroxy-5-methyl
-4-isoxazole propionic acid)
mGluR5: metabotropic glutamate
receptors
13Liu et al., Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell, 172, 979–992, February 22, 2018. https://
doi.org/10.1016/j.cell.2018.01.012
Targeted demethylation of the CGG expansion [by dCas9-Tet1/single guide RNA (sgRNA)] switched the heterochromatin
status of the upstream FMR1 promoter to an active chromatin state, restoring a persistent expression of FMR1 in FXS iPSCs.
Neurons derived from methylation-edited FXS iPSCs rescued the electrophysiological abnormalities and restored a wild-type
phenotype upon the mutant neurons.
FMR1 expression in edited neurons was maintained in vivo after engrafting (innesto) into the mouse brain. Finally,
demethylation of the CGG repeats in post-mitotic FXS neurons also reactivated FMR1.
Da questi dati si mostra che la demetilazione dell’espansione di CGG (che aveva portato alla non-trascrizione) è sufficiente per
la riattivazione di FMR1, suggerendo delle potenziali strategie terapeutiche per FXS.
iPSCs: induced Pluripotent Stem Cells
Graphical Abstract
Highlights
• Targeted demethylation of CGG repeats by dCas9-Tet1
reactivates FMR1 in FXS cells
• Demethylation of CGG repeats induces an active
chromatin status for FMR1 promoter
• Methylation-edited FXS neurons behave similarly as
wild-type neurons
In Brief
Rescue of fragile X syndrome neurons by CRISPR-
mediated DNA methylation editing of the FMR1 gene.MUTAZIONE DINAMICA
tipo di mutazione che cambia da generazione a generazione e nell’individuo durante le
fasi precoci dell’embriogenesi
Individui portatori
Individui malati
15MUTAZIONI DINAMICHE e PATOLOGIE UMANE
16DIAGNOSI
Espansione delle triplette
➛ Slittamento replicativo
➛ Scambio tra cromatidi fratelli (SCE)
➛ Errori di crossing-over durante la meiosi/
riparo
➛ Conversione Genica:
scambio non reciproco fra sequenze per
effetto del quale una sequenza (accettrice)
diventa identica alla sequenza donatrice che
rimane inalterata.
Può avvenire fra sequenze alleliche
(conversione genica interallelica) o non alleliche
(conversione genica interlocus)
F
R 17Slittamento replicativo
18Sister chromatid exchange (SCE)
is the exchange of genetic material between two identical sister chromatids.
A) Mechanism for occurrence of SCE when the leading strand
of a replication fork encounters a SSB or gap.
Resolution Holliday
=junction SCE
processing broken duplex
creates a 3′ ss-tail. Rad51 mediates strand invasion. Resolution in the orientation
(purple arrowheads) not produce SCE.
Steps 1 and 2: fork approaches a SSB.
Step 3: fork breaks.
Step 4: repair synthesis occurs at the gap in the unbroken chromatid. The curved black arrow signifies
a conformational change to facilitate visualization of subsequent events.
Step 5: processing of the broken duplex creates a 3′ single-strand tail.
Step 6: Rad51 mediates strand invasion.
Step 7: resolution of the Holliday junction in the orientation shown by the green arrows results in SCE,
as illustrated by the red/blue color junctions in the new “parental” strands. Resolution in the
orientation shown by the purple arrowheads would not produce an SCE.
Step 8: the replication fork is restored.
Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 616(1–2):11-23, 2007.
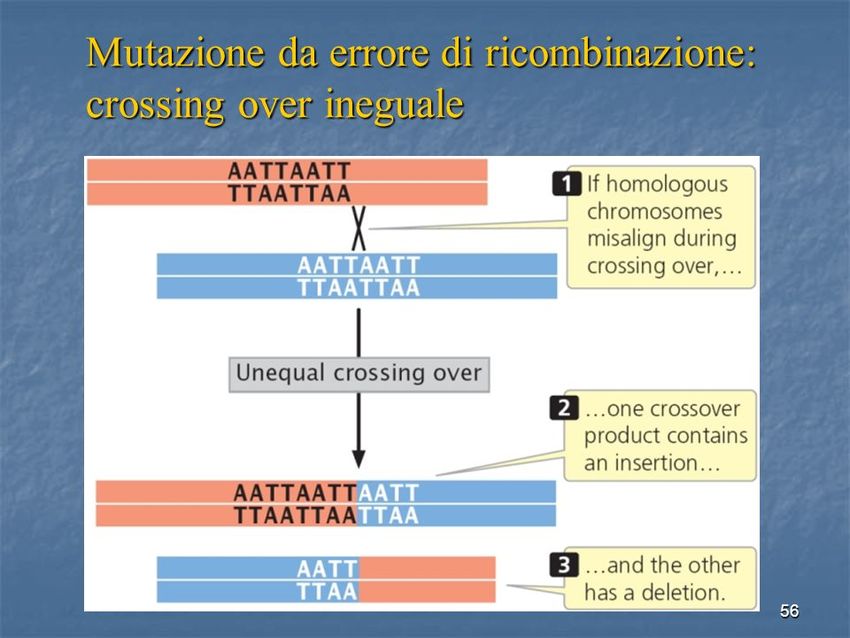
http://dx.doi.org/10.1016/j.mrfmmm.2006.11.017 19Crossing over ineguale
20Conversione genica
21Come avviene l’espansione?
• Durante la meiosi, in particolare quella femminile
Sembra essere presente una selezione negativa nella conversione tra stadio pre-mutato a stadio mutato nella
gametogenesi maschile
La premutazione espande in mutazione piena quando è trasmessa da una femmina portatrice ma non da un maschio
portatore perché l’espansione della premutazione in mutazione piena avviene solo durante la meiosi dei gameti
femminili perciò un mnaschio portatore ha solo alleli con premutazione nel suo sperma
• le figlie di un maschio portatore non sono a rischio di manifestare il fenotipo dell’X fragile
• quasi tutti i maschi con mutazione piena presentano i sintomi della malattia mentre circa la metà delle femmine
presentano ritardo mentale di grado variabile
GENETICA DELLA SINDROME DELL’X FRAGILE
La sindrome dell’X Fragile è una sindrome legata all’X
a trasmissione dominante con penetranza ridotta ( 80% - 30% )
Differenza tra e Variazioni nell’espressione del fenotipo
associato
Espressività Variabile, diventando più
Frequenza grave nelle generazioni successive
1/4000 maschi
1/6000 femmine
22Nelle cellule somatiche dei pazienti X-FRA l’instabilità mitotica delle ripetizioni della mutazione piena del gene FMR1
causa delle espansioni più corte o più lunghe, perciò tutti i pazienti sono
MOSAICI SOMATICI
• mosaicismo di lunghezza n° di ripetizioni mutazione piena - premutazione)
• mosaicismo di metilazione stato di metilazione (mutazione piena)
Differenze tra maschi e femmine
• I maschi che presentano uno stato di mutazione piena manifestano il fenotipo
• Le femmine con mutazione piena presentano un fenotipo più lieve…
46,XX un allele produce la proteina WT - Inattivazione casuale dell X
• Basi molecolari della fragilità
Strutture secondarie inusuali Esclusione dai complessi istonici
Blocco replicativo Difetti di condensazione
23L’interferenza con il processo di replicazione induce a livello delle regioni fragili un’ulteriore rallentamento della
replicazione causando in tali siti rotture e sottocondensazioni
24Altri siti fragili rari…
25Caratteristiche dei siti fragili rari clonati
Locus Agente Ripetizione Numero ripetizioni
inducente
Normale Premutazione Mutazione
piena
FRAXA
(Xq27.3) Acido folico CCG 6-52 59-230 230-2000
GGC
FRAXE
(Xq28) Acido folico CCG 4-39 31-60 200-900
FRAXF
(Xq28) Acido folico CCG 7-40 ? 306-1000
FRA11B
(11q23.3) Acido folico CCG 11 80 100-1000
2627
Siti fragili rari BrdU sensibili
Analogo strutturale della timidina,
alogenata in posizione 5
Il suo maggiore ingombro sterico rispetto alle altre basi cambia la conformazione
della cromatina in una forma più aperta, impedendo il riconoscimento della doppia
elica da parte di numerosi enzimi, tra cui la DNA polimerasi
⇓
Tutti i siti fragili rari indotti dalla BrdU fino ad oggi caratterizzati mostrano a livello
della rottura/sottocondensazione, un’espansione di dinucleotidi AT
FRA10B
• non associato ad alcuna patologia o fenotipo
• colocalizza con un minisatellite polimorfico ricco in AT
la cui sequenza consenso è di circa 42bp, in grado di
ripiegarsi in una struttura non-B stabile
• il numero delle ripetizioni è variabile tra individui diversi:
Alleli normali a basso numero di copie (circa il 66% degli alleli
– fino a1 kb)
Alleli normali a medio numero di copie
(circa il 33% degli alleli – fino a 4 kb)
Alleli normali ad alto numero di copie
(circa l’1% degli alleli – fino a 5 kb)
FRA10B minisatellite consensus sequence 28In un processo simile a quello proposto per FRAXA il numero di copie del mini-satellite può
espandere ad un livello critico (sopra le 5Kb) manifestandosi a livello citogenetico come sito fragile
Interferenza con la replicazione
Difetti di condensazione
Siti fragili rari Distamicina A sensibili
Antibiotico oligopeptidico in grado di legarsi a sequenze ricche in A/T
esternamente al solco minore della doppia elica. Il legame della distamicina A
al DNA ne altera la normale conformazione, interferendo con l’attività della
DNA polimerasi.
⇓
Anche i siti fragili rari indotti dalla distamicina A fino ad oggi caratterizzati mostrano
a livello della rottura/sottocondensazione, un’espansione di dinucleotidi AT
29FRA16B
• non associato ad alcuna patologia o fenotipo
• colocalizza con un minisatellite polimorfico ricco in AT la cui
sequenza consenso è di 33bp, in grado di
ripiegarsi in una struttura non-B stabile
• il numero delle ripetizioni è variabile tra individui diversi:
Alleli normali (circa 7-12 copie)
Alleli che esprimono il sito fragile
(circa 2000 copie)
Quindi…
• I siti fragili rari presentano un sito specifico di rottura a livello dell’espansione di tri- e di-
nucleotidi
• L’espansione è responsabile delle rotture o sottocondensazioni in quanto interferisce con il
processo di replicazione del DNA e con la formazione delle strutture di ordine superiore della
cromatina
30Puoi anche leggere

















































