Colture cellulari in dispositivi microfluidici
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Colture cellulari in dispositivi microfluidici
Relazione di:
Giovanni Marco Zaccaria
Matricola: 676687
Tutor Universitario:
Prof. Gianfranco Beniamino Fiore
Tutor Aziendale:
Ing. Matteo Moretti
Azienda:
Istituto Ortopedico Galeazzi SPA
Anno Accademico 2009-2010
COLTURE CELLULARI
IN DISPOSITIVI MICROFLUIDICI
INDICE
1. SOMMARIO
2. KEYWORDS
3. BASE DI PARTENZA ED
OBIETTIVI
4. ATTIVITA’ SVOLTE
5. LABORATORIO DI CULTURE
CELLULARI
6. LABORATORIO DI
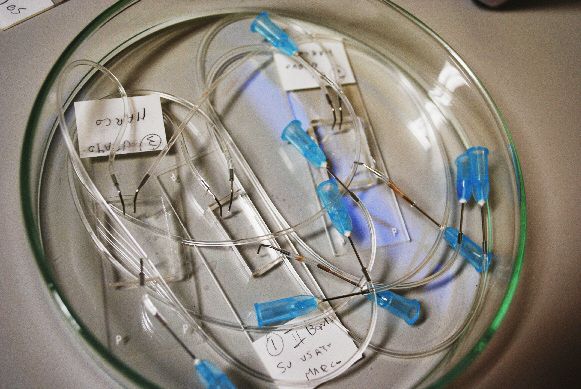
fig.1 Dispositivo microfluidico costruito nel
corso del tirocinio utilizzato durante una MICROFLUIDICA
cultura cellulare.
7. BIBLIOGRAFIA
1.SOMMARIO
Il tirocinio proposto, è stato svolto presso l’Istituto Ortopedico Galeazzi nei laboratori
di ricerca in Ingegneria dei Tessuti. Fortemente interdisciplinare ed impegnativo, ha
permesso di apprendere i principi fondamentali di culture cellulari e parallelamente, di
affrontare uno studio sia sperimentale, mirato alla produzione di dispositivi microfluidici,
che teorico, tramite approfondite ricerche bibliografiche, sullo stato dell’arte raggiunto
nel campo della microfluidica. Successivamente le attività sono confluite, raggiungendo
gli obiettivi prefissati, nella realizzazione di culture cellulari in dispositivi microfluidici (fig.1)
(fig.2). Tali attività sono state precedute da un primo periodo, di grande valore formativo,
volto alla graduale introduzione ad un ambiente di lavoro articolato e complesso, come
può essere quello, di un moderno laboratorio di ricerca. Inoltre affiancare il lavoro di
ricercatori professionisti, è un’esperienza non secondaria in grado di fornire un
importante orientamento e spunto di riflessione sul proprio futuro professionale.
2.KEYWORDS
[MICROFLUIDICA] [COLTURE CELLULARI] [BIOCHIP] [PDMS] [CONDROCITI]
[MICROFLUIDIC] [CELL COLTURE] [BIOCHIP] [PDMS] [CONDROCYTES]
2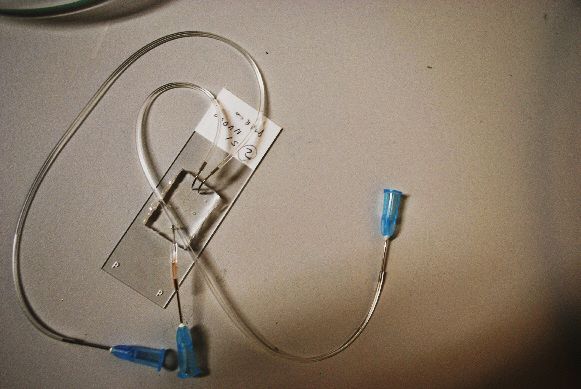
3.BASE DI PARTENZA ED OBIETTIVI
Data la natura innovativa e sperimentale del tirocinio, l’esperienza si è configurata
come totalmente complementare a quanto appreso durante gli insegnamenti del triennio
del Corso di Laurea in Ing. Biomedica.
Eccezion fatta per fondamentali nozioni di biologia e di meccanica dei fluidi, ciò che del
background, si è rivelato più efficace, è stato la “forma mentis” propria dell’ingegneria. Si
è potuto così mettere in pratica, quanto appreso in modo puramente teorico, nel corso di
Ingegneria dei Tessuti; approfondendo e dotando di maggiore organicità, la conoscenza
previa, in particolare venendo a conoscenza di una notevole quantità di tecniche e
semantiche (nonché di materiali, sostanze e strumenti) di figure professionali legate
strettamente al campo della biologia e delle biotecnologie.
Infine essendosi rivelato il primo approccio al mondo della ricerca scientifica, si è
provveduto a colmare una serie di lacune metodologiche e formative, essenziali per
potersi muovere nell’ambiente di lavoro in sicurezza ed autonomia e per poter utilizzare
correttamente e provvedere alla manutenzione ordinaria, di attrezzature e
strumentazione proprie di un laboratorio di ricerca.
Obiettivo principale delle attività è stato quindi il raggiungimento di una formazione
teorico-pratica sulla microfluidica applicata alle colture cellulari, colmando gradualmente
le carenze di operatività in campo biologico, proprie di specifiche figure professionali o
comunque altamente specializzate e contemporaneamente, approfondendo lo studio
della microfluidica (fig.3).
Fig.2 dettaglio di una semina realizzata in un biochip
3
4.ATTIVITA’ SVOLTE
Colture cellulari in
dispositivi microfluidici
Colture Dispositivi
cellulari microfluidici
Strumentazione, metodi e Strumentazione, metodi e
materiali materiali
Lavorare Preparare
sotto cappa il PDMS
Prelievo di cartilagine da Produzione
biopsie di biochip
Digestione della Il bonding
cartilagine
Espansione di condrociti Assemblaggio e
sterilizzazione
La conta delle cellule Sistema
di perfusione
Tecniche di Semina, adesione,
congelamento imagining.
fig.3 In questo grafico vengono idealmente riassunte tutte le attività
Pellet svolte. Esse si sviluppano lungo due tronconi convergenti
all’obiettivo interdiscipilinare del tirocinio.
4
5.LABORATORIO DI COLTURE CELLULARI
5.1 IL PROBLEMA DELLA STERILITÀ
L’ingresso indesiderato nel terreno di coltura di microrganismi è
dannoso: questi competono con le cellule in coltura (che
generalmente hanno ritmo di crescita inferiore) e possono
Fig. 4 operatore al lavoro sotto cappa.
secernere sostanze tossiche, compromettendo la coltura.
Tipici contaminanti sono batteri, micoplasmi, lieviti, muffe. Al fine di mantenere la sterilità
per la coltura cellulare, il laboratorio di colture cellulari deve essere esclusivo: occorre
lavorare sotto cappe a flusso laminare (fig. 4), secondo precisi protocolli. Tutti i materiali
utilizzati per la manipolazione (pipette, falcon, flask, petri, ecc..) sono sterili e di norma
monouso. Inoltre si procede con la sterilizzazione dei materiali, per i ferri chirurgici si è
ricorsi ad autoclave (a calore umido, la sterilità è garantita a una temperatura inferiore
rispetto al secco), infine per le sostanze organiche, alla Filtrazione con filtri da 0.22 μm.
La cappa a flusso laminare [1] (fig.5) è dotata di un
ventilatore che aspira una certa portata di aria dal piano di
lavoro, la filtra attraverso filtri assoluti e la invia
nuovamente nella zona di lavoro. Per evitare che si
formino vortici o zone di ricircolo il flusso di aria viene
mantenuto in condizioni laminari e viene aspirata
all’ingresso della cappa l’aria esterna verso il ventilatore. Il
flusso laminare è un flusso d'aria unidirezionale formato da
filetti d'aria sterili paralleli che si muovono alla medesima
velocità in tutti i punti, così da creare una corrente d'aria
Fig. 5 Cappa a flusso laminare. omogenea senza turbolenze. In un ambiente sterile così
ottenuto ogni contaminante libero nella zona di lavoro viene trascinato lontano da un fronte
di aria sterile. Infine è dotata di una zona di accesso frontale per il prelievo e
posizionamento di materiale, e di un piano di lavoro contenente scanalature in modo che
l’aria aspirata dal ventilatore possa essere rimessa in circolo.
5
5.2 STRUMENTAZIONE, METODI E MATERIALI
I sistemi di coltura [1] in cui vengono processati i campioni e coltivate
le cellule sono le flask (T150, T75 e T25), in plastica trasparente
(fig.6) ed internamente sterili; inoltre possono avere un tappo
ventilato, cioè con un filtro che permette gli scambi gassosi e
Fig. 6 Rappresentazione di contemporaneamente garantisce la sterilità della coltura.
due flask di dimensioni
Questi contenitori hanno solitamente una superficie inferiore liscia
differenti.
per permettere di osservare il preparato al microscopio. La
superficie inferiore di questi contenitori è solitamente trattata per esporre cariche
superficiali che favoriscono l’adesione delle cellule, ed il numero con le quali vengono
identificate, rappresenta l’area di tale superficie espressa in cm2 .
L’incubatore a CO2 [1] è un’attrezzatura di precisione per il
mantenimento delle cellule in coltura (fig.7): è costituito da una
camera chiusa al cui interno sono ricreate, nei limiti del possibile, le
condizioni fisiologiche dei tessuti animali in cui le cellule vivevano
prima dell’isolamento.
Fig. 7 Interno di un I parametri che vanno tenuti sotto controllo sono:
incubatore a CO2.
Temperatura: il controllo della temperatura (che deve essere mantenuta costante a
37°C) viene assicurato da un termostato che agisce su una resistenza; un sistema di
ventole per la circolazione forzata dell’aria garantisce che la temperatura sia
uniforme in tutti i punti della camera.
Concentrazione di CO2: l’aria atmosferica è miscelata con il 5% di CO2.
pH: deve essere di 7,4; dipende dal riequilibrio tra lo ione bicarbonato contenuto nel
terreno di coltura e la CO2.
Umidità: l’ambiente deve essere saturo; allo scopo nell’incubatore è presente un
vassoio di umidificazione contenente acqua.
6
5.3 TECNICA DI PIPETTAGGIO
Le pipette (fig. 8) sono determinanti per la qualità del risultato analitico in
laboratorio, permettono misurazioni precise e riproducibili di liquidi, in sicurezza
e senza rischi di contaminazione.
• Settare la pipetta sul volume da pipettare
• Premere il pulsante di comando: il pistone elimina parte del volume morto
• Immergere il puntale nel liquido da prelevare
Fig. 8 Schema di
• Rilasciare la pressione sul pulsante di comando per consentire una micropipetta
l’aspirazione del liquido
• Sollevare il puntale dal liquido facendolo scorrere lungo la parete del recipiente ed
appoggiare la punta del puntale contro la parete del recipiente ricevente
• Premere il pulsante di comando fino al primo fermo; continuare la pressione fino al
secondo fermo per far defluire i residui di liquido rimasti.
• Premere l’espulsore per far staccare il puntale dalla micropipetta.
PUNTALI
I puntali sono una parte fondamentale del sistema di pipettaggio. I puntali con filtro hanno
lo scopo di bloccare e proteggere sia la micropipetta che il campione, evitando le cross-
contaminazioni. Devono essere conformi ai seguenti criteri:
• Esenti da contaminazione biologica e chimica
• Fabbricati in materiale resistente agli attacchi chimici
• Puliti, esenti da polvere
• Devono agganciarsi perfettamente alla punta della micropipetta
• Devono essere stampati con superfici lisce e senza alcun difetto
FALCON: sono provette a fondo conico (fig.9)con capienza di 15 e 50 ml
utilizzate per riporre i reagenti liquidi e per la centrifugazione delle
sospensioni cellulari.
Fig. 9 Provette falcon da
50 e da 15 ml.
7
5.4 SOSTANZE PIU’ UTILIZZATE
MEM (Minimum Essential Medium): è il terreno di coltura.
FBS (Siero Fetale Bovino): contiene numerosi fattori di crescita (non contenuti nel terreno
di coltura) utili per la
crescita delle cellule.
PBS (Phosphate buffered saline) è una soluzione salina che contiene cloruro di sodio,
sodio fosfato e potassio fosfato. Il tampone aiuta a mantenere costante il pH.
La concentrazione salina è la medesima del corpo umano (cioè è isotonica). Il PBS ha
molti usi soprattutto perché è isotonico e non tossico per le cellule, infatti è anche usato
come soluzione di lavaggio per le cellule. Può essere usato per diluire le sostanze.
TRIPSINA: enzima proteolitico che rompe i legami peptidici delle proteine extracellulari
che costituiscono il substrato su cui le cellule sono ancorate, nel nostro caso è stata
utilizzata, per staccare le cellule dalla plastica della flask: la temperatura ottimale per il suo
funzionamento è 37°C ma funziona anche a temperatura ambiente. In questo caso
occorre lasciar agire per un tempo più lungo.
TRYPAN BLUE: è un colorante usato durante la conta cellulare e che diffonde solo nelle
cellule la cui permeabilità di membrana è fortemente danneggiata, il che implica che le
cellule che appariranno colorate in blu sono cellule da considerare morte. Il Trypan Blue
ha effetto tossico, quindi va aggiunto solo poco prima di effettuare la conta con il
microscopio ottico.
8
5.5 LA DIGESTIONE DELLA CARTILLAGINE
Il materiale da cui vengono isolate e messe in coltura le
cellule proviene solitamente da un organismo vivente
[1]. Nel nostro caso, il materiale biologico è stato fornito
da campioni di tessuti provenienti da pazienti prelevati in
sterilità durante operazioni chirurgiche mirate alla
rimozione e sostituzione con protesi, di teste femorali
(fig. 10).
fig.10 Rappresentazione dell’estremità
superiore di un femore, si può notare la testa,
da cui originano le biopsie di cartilagine
utilizzate.
Una volta prelevato il campione di tessuto si deve procedere a separarne le cellule dalla
matrice circostante. I processi di separazione sono di due tipi differenti: meccanico ed
enzimatico, eseguiti in sequenza. Per isolare le cellule si procedere a frammentare il
campione servendosi di un bisturi (fig. 11), ciò per evitare che la mancata ossigenazione
provochi la morte delle cellule. Con il processo di separazione enzimatica la digestione
dell’ECM viene effettuata con un apposito enzima, la collagenase, che demolendo la
matrice extracellulare e le giunzioni intercellulari che mantengono unite le cellule, permette
di disperdere le singole cellule nella soluzione. È necessario bloccare il processo di
digestione dopo 22h, per evitare che gli enzimi danneggino anche le cellule.
La sospensione cellulare omogenea così ottenuta è messa in “coltura”, cioè viene
trasferita in contenitori appositi contenenti miscele di sostanze inorganiche ed organiche
necessarie al sostentamento delle cellule.
fig.11 Bisturi a lama monouso, la lama si inserisce fino ad uno scatto che ne
segnala il corretto posizionamento. Il corpo bisturi a fine lavoro verrà sterilizzato.
9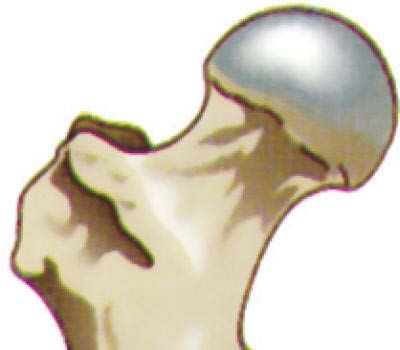
5.6 COLTURE PRIMARIE
Una coltura primaria viene generata isolando cellule direttamente a partire da tessuti o
organi, le cellule isolate in questo modo riflettono meglio le attività biochimiche delle cellule
in vivo, tuttavia hanno una vita limitata e, per la realizzazione di progetti a lunga scadenza,
è necessario prevedere diversi isolamenti.
Una volta isolate da un tessuto, dalla sospensione cellulare ottenuta si
ottiene una coltura primaria. Le cellule della coltura primaria
aderiscono alla piastra e crescono fino a coprire tutta la superficie
disponibile, ossia fino a quando arrivano a confluenza. A questo punto
vengono rimosse dalla piastra (fig. 12) di coltura e sistemate in nuove
piastre a bassa densità allo scopo di ottenere colture secondarie.
fig.12 Operare all’interno di una flask richiede molta attenzione,
bisogna sempre inclinarla per evitare contaminazioni provenienti
dalla mano.
Da una fiasca contenente cellule a confluenza:
• Scartare il terreno di coltura;
• Lavare con PBS;
• Aggiungere tripsina e lasciar agire la tripsina per circa 5 min a
37°C in incubatore;
• In questo tempo preparare un tubo contenente CM (fig. 13),
riportando i dati della coltura;
• Trascorsi i 5 minuti, verificare che le cellule si siano staccate;
Fig. 13 Complete
quindi prelevare le cellule, metterle nel tubo con CM e Medium (CM)
centrifugare;
450ml Dulbecco’s
• Scartare il sopranatante (sul fondo del tubo è visibile il pellet, modified Eagle’s
composto dalle cellule sedimentate); medium (DMEM)
• Aggiungere al sedimento CM in modo da risospendere le
50ml Fetal bovine serum
cellule, fino a raggiungere il volume desiderato. (FBS)
• Prelevare un piccolo volume di sospensione cellulare per
5 ml HEPES (1M):
effettuare la conta delle cellule
• Seminare in flask, il volume di sospensione cellulare da 5 ml Sodium pyruvate
seminare dipende dalla superficie di crescita della fiasca e solution
dalla densità desiderata. 5 ml PSG
105.7 OSSERVAZIONE DI CELLULE AL MICROSCOPIO
La microscopia è una risorsa indispensabile per l’osservazione e
lo studio di campioni di natura diversa (fig. 13). Un uso tipico
della microscopia consiste nell’osservazione di cellule eucariote
che crescono adese alla superficie di coltura. Le cellule tenute in
fig.13 grazie all’ingrandimento su queste condizioni sono in genere molto sottili e sono
di una cultura cellulare, possiamo trasparenti alla luce. Per l’osservazione, vengono spesso
monitorarne l’adesione.
utilizzate metodiche che permettono di superare questo
problema, aumentando il contrasto, attraverso l’uso di dispositivi capaci di generare
contrasto di fase o l’introduzione di coloranti che assorbono nello spettro del visibile.
In un microscopio ottico (fig. 14) la luce emessa da una lampada
a incandescenza viene concentrata, attraverso le lenti di un
condensatore, che focalizzano i raggi emessi sul piano del
campione. La luce che fuoriesce dal campione, utilizzando una
combinazione di due lenti, l’obiettivo e l’oculare, viene focalizzata
direttamente sull’occhio dell’osservatore, o su un sistema di
registrazione costituito da una pellicola fotosensibile o da una
telecamera. Per garantire la qualità dell’immagine è necessario
fig.14 microscopio ottico.
utilizzare lenti di buona qualità e una meccanica molto stabile,
che riduca le vibrazioni. In assenza di altre limitazioni, la massima risoluzione ottenibile
con un microscopio ottico è determinata dalla lunghezza d’onda della luce visibile, che
pone un limite teorico di 0.2 µm.
115.8 LA CONTA CELLULARE
Per contare le cellule con la camera di Burker (fig. 15), si
agita bene la sospensione cellulare usando una pipetta di
plastica sterile, evitando la formazione di bolle e schiuma.
Quindi si prelevano circa 0,2 ml di sospensione che
vengono messi un una provetta Eppendorf non sterile.
fig.15 Camera di Burker
A questo punto la provetta Eppendorf (fig. 16) viene portata fuori cappa
sterile e le cellule vengono contate. Per valutare la mortalità cellulare, le cellule
vengono colorate diluendo il campione 1:2 ( o anche 1:10 per campioni con elevata
densità cellulare) con soluzione di Trypan Blue al’1% in PBS. Si deve preparare la
diluizione con il Trypan Blue con precisione perché è necessario conoscere
esattamente il fattore di diluizione per risalire poi alla concentrazione di cellule
presente nel campione analizzato. fig.16 Disegno di una
provetta Eppendorf
Procedimento:
• Controllare che la Burker sia pulita e montata correttamente
• Prelevare una piccola quantità del campione da contare e
appoggiare la punta della pipetta contro il bordo del vetrino
copri oggetto. Lasciare fuoriuscire una goccia del campione
fig.17 Struttura di una
che per capillarità si diffonderà nella camera di Burker. camera di Burker
• Contare le cellule contenute nei quadrati (fig. 17) disposti lungo le diagonali della
camera e farne la media.
• Per conoscere il numero di cellule presenti nella sospensione di partenza, applicare
la seguente formula:
N° di cell. totali = Media * Fattore di camera* diluizione trypan blue * ml della sosp. iniziale
Con fattore di camera = 10.000
125.9 IL CONGELAMENTO
Portare avanti delle colture non necessarie e' un inutile
spreco di tempo e di materiale, cioè di denaro. Inoltre continue
espansioni portano ad aberrazioni genetiche e, specialmente
nel caso di cellule normali, non trasformate, ad un
invecchiamento della coltura, che può rispondere diversamente
ai trattamenti e alle indagini degli studiosi, ed e' bene quindi
avere degli stock conservati. Le cellule devono essere
fig.18 Dispositivi di protezione congelate solo quando sono nella fase esponenziale di crescita
durante l’utilizzo di azoto liquido. e quando sono in buona salute. E' inoltre importante controllare
attentamente che non vi siano contaminazioni. Per le cellule eucariote, il sistema preferito e' il
congelamento in azoto liquido, basato sul principio che le sole reazioni che avvengono a -
176° (temperatura dell'azoto liquido) sono le ionizzazioni dovute alla radiazione di fondo,
reazioni che peraltro sono piuttosto rare. Le reazioni enzimatiche cessano a -136°C circa.
NORME DI SICUREZZA : Prima di lavorare con l'azoto liquido, e' bene chiedere l'assistenza
di personale esperto. L'azoto liquido infatti causa bruciature dovute alla sua temperatura
estremamente bassa. ll materiale congelato e l'azoto liquido vanno manipolati indossando i
cosiddetti dispositivi di protezione individuali e cioè i guanti appositi (fig. 18) e la maschera di
protezione. Inoltre bisogna prestare attenzione all' l'azoto che può entrare nelle ampolle
congelate, e con il riscaldamento passa allo stato di vapore e quindi causare l'esplosione
delle ampolle stesse.
CRIOPROTETTIVO: Il congelamento deve mantenere elevata la vitalità cellulare per
tutto il periodo di congelamento. E' molto importante sia la velocità di congelamento che di
scongelamento. Per quanto riguarda il congelamento bisogna ricordare che il congelamento
rapido porta alla formazione di cristalli di ghiaccio all'interno della cellula, cristalli che, al
momento dello scongelamento, portano alla rottura della membrana plasmatica. Basse
velocità di congelamento portano invece alla deposizione di ghiaccio nell'ambiente
extracellulare, a condizione però che la concentrazione interna di soluti sia sufficientemente
alta da rendere la concentrazione dell'acqua intracellulare minore di quella extracellulare.
Questo processo viene di solito controllato aggiungendo al terreno di congelamento un
agente crioprotettivo permeabile, il dimetilsolfossido (DMSO).
13Procedimento:
Le cellule devono essere tripsinizzate se sono
adese, contate e centrifugate; le cellule aderenti
vengono di solito congelate ad una concentrazione di
circa 5 x 106 cell/ml. Il nostro protocollo prevede che
le cellule siano centrifugate, venga scartato tutto il
sopranatante e il pellet risospeso nel terreno da
fig.19 Criotubes pronti al congelamento. congelamento, a questo punto si mescola bene la
sospensione cellulare e aliquote da 1 ml della sospensione cellulare vengono poste in
provette apposite dette criotubes (fig. 19), tutta la procedura va fatta in sterilità. I tappi dei
criotubes non devono essere chiusi stringendoli al massimo, dato che questo può causare
una distorsione della guarnizione e permettere l'entrata di azoto liquido nella provetta.
Dobbiamo tra l'altro ricordarci che l'azoto, anche se molto freddo, non e' sterile e l'entrata di
azoto nella provetta puo' essere molto pericolosa.
Il congelamento ottimale viene effettuato a velocità costante nei vapori di azoto che si
formano nel criocontenitore: la velocità di raffreddamento dovrebbe essere di 1 °C/min, e
viene di solito ottenuta con un "tappo speciale" che permette di abbassare a velocità
costante i campioni nei vapori, costituito da una scatola di polistirolo espanso immersa in
isopropanolo. Ogni scatola dovrebbe alloggiare 10-
20 criotubes che devono rimanere in piedi per
evitare che il materiale si congeli nel tappo (questo
può essere pericoloso per le contaminazioni). La
scatola viene chiusa ermeticamente e messa in un
congelatore a -80°C (fig. 20). Le cellule in queste
condizioni raggiungono la temperatura dei vapori
dell'azoto liquido (-80°C) in circa 3 ore. A questo
punto si mettono le cellule in azoto liquido. Sarebbe
bene non lasciare a lungo le cellule a -80 °C.
fig.20 Congelatore verticale a -80°C
14SCONGELAMENTO
I criotubes devono essere prelevate dal contenitore di azoto con molta precauzione, lo
scongelamento deve essere fatto molto velocemente, quindi le ampolle vengono passate con
l'alcool al 70% e messe sotto cappa. Il contenuto della ampolla (1 ml) deve ora essere diluito
con 7 ml di terreno completo e centrifugato per rimuovere il terreno di congelamento. La
diluizione può essere un punto critico della procedura, dato che si può verificare un forte
shock osmotico al momento della diluizione con il terreno normale, per avere una diluizione
graduale si può aspirare la sospensione appena scongelata in una pipetta da 10 ml e di
prelevare con la stessa pipetta i 7 ml di terreno. Per non contaminare tutta la bottiglia di
terreno, i 7 ml da utilizzare verranno precedentemente messi in una provetta, eventualmente
la stessa provetta in cui poi centrifugheremo le cellule. Le cellule vengono centrifugate, il
sopranatante eliminato e le cellule risospese in 10 ml di terreno completo. Un campione
viene prelevato per contarlo e per valutarne la vitalità.
Le cellule vengono inoculate in fiasca ( o in altro contenitore) e dopo 24 ore la coltura viene
osservata al microscopio e se sono cellule sospese possiamo ricontarle. Se sono cellule
adese, dopo 24 ore devono essersi attaccate al substrato. Se le cellule non si sono attaccate
o sono tutte morte ovviamente si scartano e si deve procedere al controllo di altri criovials per
valutare se l'intero batch e' morto, il che indica che la procedura di congelamento e' stata
fatta in modo sbagliato.
Le diverse linee cellulari manifestano una diversa capacità di ripresa dopo il
congelamento: in alcuni casi si ha un'altissima mortalità e le cellule hanno bisogno anche di
3-4 giorni per riprendersi completamente, altre cellule si riprendono immediata mente.
155.10 PELLET
I condrociti in cultura convenzionale (bidimensionale) possiedono un fenotipo altamente
instabile. In quanto in cultura bidimensionale, i condrociti subiscono
una dedifferenziazione , trasformandosi in fibroblasti secernendo
collagene di tipo I nella matrice, ma perdendo l'espressione sia di
collagene di tipo II che delle core protein (aggrecano). La densità
cellulare è uno dei requisiti critici per stabilizzare il fenotipo dei
condrociti. Quando i condrociti sono coltivati ad alta densità (105 cm-2)
in flask di cultura, il loro fenotipo non cambia.
fig.21 Eppendorf contenente
un pellet realizzato durante il
tirocinio.
Le cellule quindi vengono spesso coltivate ad alta densità per promuovere la
differenziazione condrogenica e mantenerne il fenotipo, compattando i condrociti in una
micromassa sferica, il pellet [3]. Tale sistema di cultura pellet (fig. 21) permette la crescita
dei condrociti in ambiente tridimensionale (senza ricorrere a scaffold), che determina il
mantenimento delle caratteristiche di cellule differenziate come nella cartilagine in vivo,
consentendo un mantenimento in coltura per periodi di tempo superiori a quanto è
possibile per i condrociti in monostrato, che iniziano in 7-8 giorni un processo di
differenziamento.
166.LABORATORIO DI MICROFLUIDICA
L’uso dei dispositivi microfluidici [4] per la ricerca biomedica ha una serie di vantaggi
significativi. Primo, il volume di fluidi all’interno di questi canali è estremamente ridotto,
solitamente nell’ambito dei microlitri. Ciò è di particolare importanza per un utilizzo
efficiente di reagenti costosi.
Questi dispositivi rendono inoltre possibile eseguire l’analisi in meno tempo e con
velocità di trasferimento superiori grazie alla riduzione delle distanze.
Inoltre, durante l’esecuzione di più dosaggi in parallelo, ciascun processo di un dosaggio
può essere manipolato passo per passo tramite il controllo di un computer, consentendo
di lavorare con maggiore efficacia. Tale accuratezza poi, combinata con rese superiori,
determina una riduzione degli scarti. I risvolti positivi di questo elemento non consistono
nella sola riduzione dei costi ma anche nella salvaguardia dell’ ambiente, poiché si tratta
spesso di sostanze chimiche dannose. Inoltre, le tecniche di fabbricazione utilizzate per
la costruzione dei dispositivi microfluidici sono relativamente economiche ed adattabili
alla realizzazione di dispositivi estremamente elaborati ed in multiplex, oltre che alla
produzione di massa.
I circuiti microfludici sono caratterizzati dalla presenza di canali che hanno dimensioni che
vanno da pochi micrometri alle centinaia di micrometri. Questi sistemi miniaturizzati
possono essere costruiti partendo da vari materiali quali vetro e polimeri.
Il flusso di liquidi nei microcanali non è influenzato dalla forza di
gravità, ma da processi di capillarità ovvero dalle interazioni
specifiche del fluido con la superficie dei canali. In questi sistemi si
ha in genere un flusso laminare, che non porta ad un efficace
mescolamento di flussi provenienti da canali diversi.
fig.22 Rappresentazione di un flusso
turbolento e di uno laminare
176.1 IL PDMS
I dispositivi impiegati nell’ambito della microfluidica vengono
spesso prodotti utilizzando due polimeri: PDMS (fig. 23)
(polydimethylsiloxane) e PMMA (polymethylmethacrylate o
fig.23 Struttura chimica del PDMS. plexiglass). Nel corso del tirocinio si è ricorsi al PDMS [2][5],
che risulta essere un materiale non solo inerte e non
tossico, ma anche biocompatibile. Inoltre è dotato di una notevole resistenza al calore,
agli attacchi chimici, all'ossidazione, è permeabile ai gas, è un ottimo isolante elettrico e
resistente all’invecchiamento; in più è otticamente trasparente sotto i 230 nm (e quindi
compatibile con la maggior parte dei metodi ottici di indagine)
Questo polimero inoltre non si lega né al vetro, né al metallo, né alla plastica in fase di
solidificazione, ma conserva maggiore aderenza sulle superfici lisce una volta solidificato,
ed è grazie a quest’ultima proprietà che il PDMS ha conosciuto questo grande successo,
infatti risulta estremamente semplice legare il dispositivo prodotto ad altre superfici. Nello
sviluppo di saggi, più soluzioni progettuali potrebbero esser prese in considerazione, la
facilità e la rapidità di prototipazione di dispositivi in PDMS rappresenta un grande punto
a favore per questo materiale. Tutte queste proprietà lo hanno reso negli anni il polimero
più utilizzato nel campo della microfluidica.
Per preparare il PDMS bisogna miscelare due componenti allo stato liquido: la base, il
Sylgard 184 silicone, in rapporto di 10:1 con il curing agent. La miscela verrà poi messa
in un contenitore sottovuoto per rimuovere qualunque bolla d’aria presente, a questo
punto sarà pronta per essere utilizzata.
18Procedimento [6]:
Completare tutti i processi sotto cappa chimica
Miscela e degas di PDMS:
- Silicone di partenza è molto appiccicoso.
fig.24 dettaglio sul processo di
Fermare con un pezzo di nastro adesivo, un foglio di preparazione di PDMS.
alluminio all’interno della cappa chimica sufficiente a
coprire tutto lo spazio di lavoro. Proteggere con un foglio di alluminio anche la bilancia.
- Mettere un bicchiere di plastica pulito, sopra la bilancia e tararla.
- Usando un cucchiaio di plastica dispensare la base del prepolimero e con una
pipetta il curing agent, in rapporto di 10:1 (agente di base:curing). Aggiungere prima la
base (fig. 24), il volume della base dovrà essere pressocché uguale al volume totale.
Aggiungere quindi il curing agent con la pipetta. Mescolare bene col cucchiaio (finché la
mistura diventa biancastra a causa delle bolle di aria).
- Mettere il bicchiere con la mistura di PDMS nell'essiccatoio ed accendere la pompa
a vuoto. Stare attenti che la mistura di PDMS non esca fuori del contenitore al momento
dell’immissione di aria. Un trucco consiste nell’aspettare che le grandi bolle arrivino in
superficie, quindi aprire il foro e immettere rapidamente aria per scoppiare le bolle.
Mantenere la mistura sotto vuoto finché non resta più nessuna bolla.
196.2 PRODUZIONE DI BIOCHIP
Il processo di produzione dei dispositivi microfluidici consiste nella modellazione del
PDMS [5][6] attraverso il master mold in modo da ottenere una copia in negativo del
master.
Il pre-prolimero viene depositato sul master mold e riscaldato per circa 2 ore a 70 C°
così da favorirne la solidificazione. A questo punto si rimuove il dispositivo in PDMS dal
master. Si procede, dunque, alla creazione dei fori (punch) per la connessione del
circuito idraulico con l’esterno.
Procedimento:
- Prestando la dovuta attenzione versare il PDMS precedentemente preparato nel
master mold, messo su un foglio di alluminio.
- Cercare di minimizzare l’introduzione di bolle, mettere quindi sotto vuoto fino alla
scomparsa di tutte le bolle d’aria.
Solidificazione del PDMS:
- PDMS solidificherà senza cottura in ~ 24 ore.
- Per velocizzare tale tempo, mettere il Master
Mold sopra l'appoggio di resina e poi in forno (fig. 25) a
70°C per ~ 2 ore (dipende dello strato di PDMS).
- Dopo la solidificazione, il dispositivo in PDMS è
stabile e può essere immagazzinato per mesi se
necessario.
- Servendosi di una spatola d’acciaio, facendo molta fig.25 forno utilizzato al fine di
attenzione a non graffiare il Master Mold, si tiri via il accelerare la solidificazione del
PDMS. PDMS.
- Coprire con del nastro adesivo le superfici maggiori del dispositivo.
- Metta il dispositivo in una Petri, poi fissarlo con sufficiente nastro adesivo, per
poterlo prendere facilmente.
206.3 BONDING
Il PDMS può essere legato in maniera irreversibile a vetro,
silicio o PDMS stesso [7]. Esponendo il PDMS ad un plasma
(fig. 26), la superficie del PDMS diviene idrofila e reattiva,
dando luogo a bonding irreversibile quando entra in contatto
con vetro, silicio, o un altro pezzo di PDMS che sono stati
fig.26 Caratteristica luce violetta emessa messi in mostra allo stesso plasma. Nel nostro
durante la funzionalizzazione delle superfici.
caso, si è fatto uso di vetrini polilisinati (fig. 27).
Questo step, e tutto il processo produttivo, termina con il bonding delle due parti
funzionalizzate.
Il dispositivo viene trattato con plasma a bassa energia (un processo che
forma legami covalenti O-Si-O), lo stesso trattamento viene effettuato sul
vetrino che farà da base del dispositivo. La funzionalizzazione delle due
facce favorirà l’adesione. Il PDMS è piuttosto idrofobico, possiede una
bassa energia superficiale che rende il materiale non reattivo, rendendo
fig.27 Vetrini utilizzati
difficile qualunque collegamento con le altre superfici. Questo
nell’assemblaggio dei contatto dovrebbe avvenire immediatamente perché il PDMS dopo
biochip.
poco, ritornerà al suo stato idrofobico.
216.4 ASSEMBLAGGIO E STERILIZZAZIONE
Fig. 28 Cannule opportunamente sagomate,
costituiscono i connettori del dispositivo. (fig. 28)
A garanzia del corretto funzionamento, prima
dell’assemblaggio si procede a testare connettori
e tuberia con acqua doppiamente distillata.
Fig. 29 I biochip assemblati verranno quindi portati sotto
cappa chimica e sterilizzati con etanolo al 70% e
risciacquati con soluzione PBS sterile. Infine
verranno pretrattati con complete medium al fine
di creare un ambiente favorevole alla coltura
cellulare. Si lasciano quindi overnight in
incubatore. (fig. 29)
Fig.30
Per proteggere da contaminazioni, si coprono i connettori
con un filtro a siringa per ogni ingresso (fig. 30).
Fig.31
Si procederà alla semina collegando ad una
pompa a doppia siringa, le due entrate con i
connettori più lunghi. Il terzo connettore invece
farà da reservoir, ossia il campione verrà iniettato
nel tappo e facendo lavorare la pompa in
aspirazione, entrerà all’interno del biochip (da qui
la brevità del tubo rispetto agli altri). (fig. 31)
Fig.32
A fine semina tutti i tubi verranno chiusi (“clampati”) per
evitare contaminazioni e per offrire ulteriore sostegno
strutturale ai filtri a siringa.(fig. 32)
226.5 IL SISTEMA DI PERFUSIONE MICROFLUIDICO
I piccoli volumi di campione usati nei biochip, l’erogazione di precisione delle soluzioni
richieste nei siti di reazione, la miscelazione di liquidi differenti, la creazione di gradienti
di concentrazione dei reagenti, il controllo della posizione dei campioni biologici, il loro
trasporto e la loro manipolazione sono tutti elementi che richiedono la presenza di un
accurato sistema di perfusione.
Comparando i valori tipici, dei vari sistemi di perfusione presenti in letteratura, si è giunti
ai seguenti, ma non sono esplicitamente limitati, intervalli:
• Volumi ridotti: da 5 nL a 100 uL
• Bassa velocità di flusso volumetrico: da 100pL/min a 10 uL /min
• Stress di trazione corrispondente: da 10-2 dyne/cm2 a 10 dyne/cm2
• Controllo della velocità e direzione di pompaggio: da 10 um/s a 1 cm/s .
Solitamente vengono usati vari assemblaggi di pompe
a spostamento positivo, incluse le pompe a siringa (fig.
33), ad infusione per pressione positiva e peristaltiche,
molti sistemi convenzionali però, lavorano ancora con
volumi notevolmente superiori di liquido, e di
conseguenza non possono fornire l’ accuratezza o, in alcuni casi, la
fig.33 pompa a siringa
velocità di pompaggio adeguata all’instaurazione del flusso nelle
strutture dei biochip con canali dal diametro compreso fra 5 e 100 um. Attualmente sono
in uso pompe a siringa capaci di instaurare una velocità di microflusso volumetrico
preciso e riproducibile nell’ ordine di 0,1 – 1 uL al minuto. Uno dei principali obiettivi è
ottenere un flusso continuo non pulsato, infatti la pressione del fluido nella pompa a
siringa cambia durante il pompaggio, solitamente controllato da un motore passo-passo.
Ciò determina un’onda pressoria che altera la velocità del flusso volumetrico e di
conseguenza, di rendere relativamente inefficienti le portate di fluido dell’ordine dei
nanolitri per minuto, tale infatti è la velocità richiesta per trasportare i liquidi all’interno
delle strutture microfluidiche dei biochip.
236.6 IMAGINING FINALE
Fig.34 Fig.35
Cellule adese all’interno del Dettaglio del foro praticato
biochip (fig. 34). con il punch (fig. 35).
.
Fig.36 Fig.37
Problematica 1: ammassi Problematica 2: bolle d’aria
cellulari (fig. 36). (fig. 37).
.
247.BIBLIOGRAFIA
[1] Mantero S., Remuzzi A., Raimondi M.T. e Ahluwalia A. Fondamenti di Ingegneria dei
Tessuti per la Medicina Rigenerativa. Pàtron Editore, Bologna, 2009
[2] M.C. Tanzi "Fondamenti di Bioingegneria Chimica: non solo biomateriali" Pitagora
Editrice, Bologna, 2006.
[3] Zijun Zhang, J. Michael McCaffery, Richard G. S. Spencer, Clair A. Francomano
Hyaline cartilage engineered by chondrocytes in pellet culture: histological,
immunohistochemical and ultrastructural analysis in comparison with cartilage
explants
[4] F. Menolascina and C. Ciminelli, Microfluidica: Teoria ed Applicazioni
[5] Samuel K. Sia George M. Whitesides, Microfluidic devices fabricated in
poly(dimethylsiloxane) for biological studies
[6] Robert White, Polydimethylsiloxane (PDMS) on SU-8 Mold SOP
[7] Robert White, PDMS-Glass Bonding via Oxygen Plasma SOP
25Puoi anche leggere

















































