Cancro - crescita e divisione cellulare incontrollate - inattivazione funzioni tissutali - morte organismo - e-learning
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Cancro -crescita e divisione cellulare incontrollate -inattivazione funzioni tissutali -morte organismo

Cancro: malattia genetica
Mutazioni
• 1. Ereditaria (predisposizione)
• 2. Acquisita (mutazioni nelle cellule
somatiche)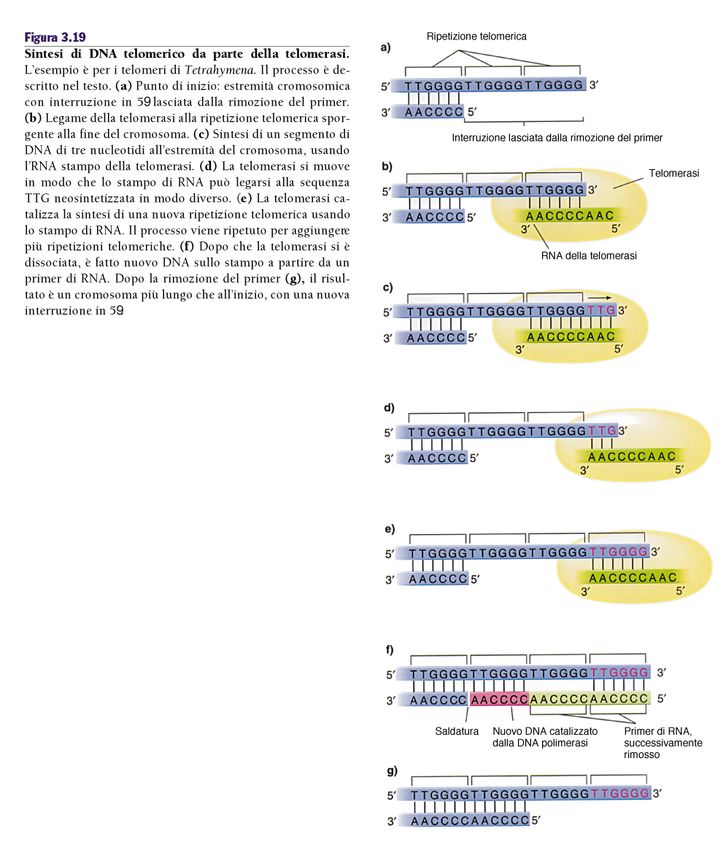
Cancro: malattia genetica
1. Ereditaria (predisposizione)
2. Acquisita (mutazioni nelle cellule somatiche)
Il caso Angelina Jolie: mastectomia per contrastare la predisposizione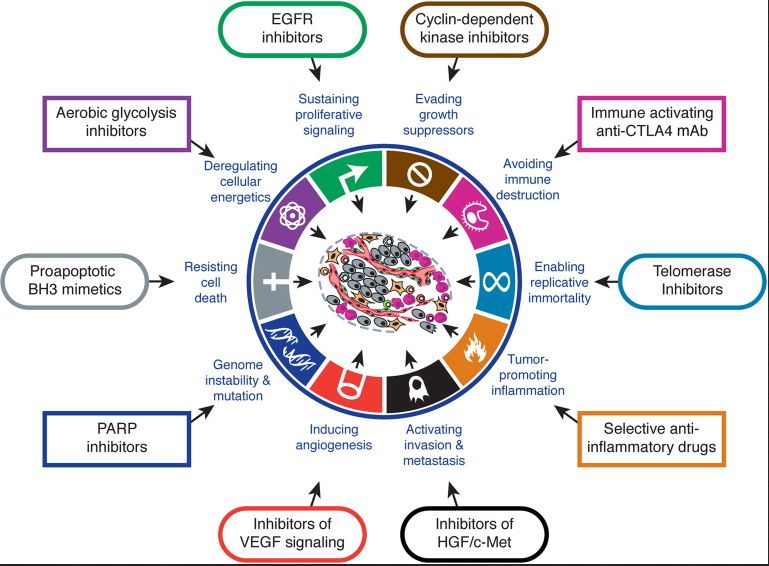
Eredità mendeliana
Eredità legata all’X
Cancro: malattia genetica
1. Ereditaria (predisposizione)
2. Acquisita (mutazioni nelle cellule somatiche)
Mutazioni: errori nella replicazione del DNA
• 1/3 di affetti da cancro durante la vita
• 104 cellule che si dividono per varie decadi di vita
• Perchè così raro?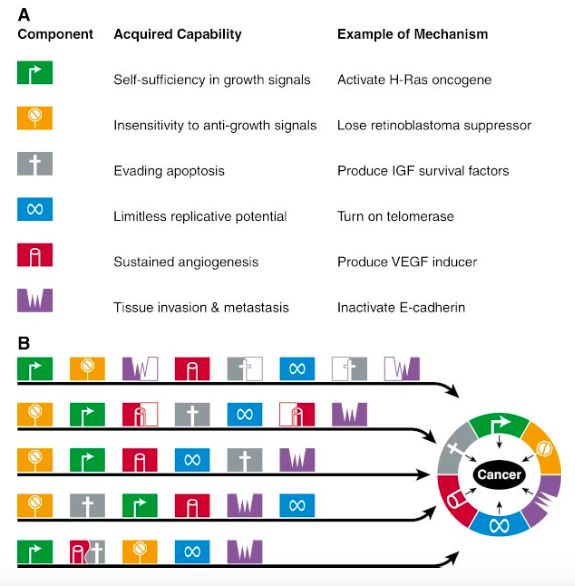
Perchè….
• Più mutazioni richieste per trasformazione tumorale
• Processo multistep analogo all’evoluzione darwiniana:
una serie di cambiamenti successivi, che favoriscono la crescita, porta alla
progressiva trasformazione di una cellula normale in tumorale
• Processi cellulari di “difesa”
• Ciclo cellulare controllato da una rete di meccanismi a feed back
• Cellule irregolari eliminate per apoptosi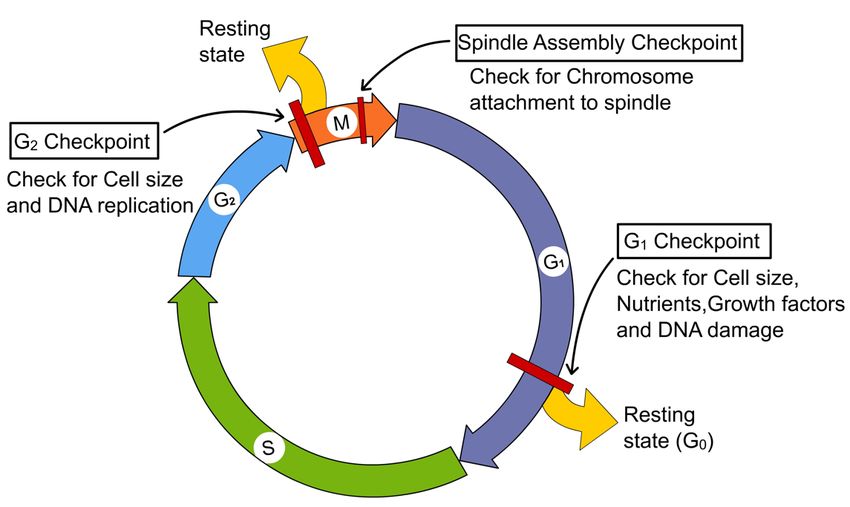
Ciclo cellulare
Transizione tra le fasi regolata da una rete di segnali chimici
specifici a sua volta collegata al ciclo cromosomico da accumulo,
modificazione e distruzione ciclici di componenti chiave
• Chinasi (proteine che fosforilano)
• evolutivamente conservate (lievito-uomo cdc2-p34)
• che per funzionare devono associarsi ad una ciclina
• CDKs (Cyclin-Dependent Kinases)
cdc2-p34 = Cdk1 = G2 M
Cicline
(16 nell’uomo)
accumulate gradualmente in interfase e distrutte in mitosi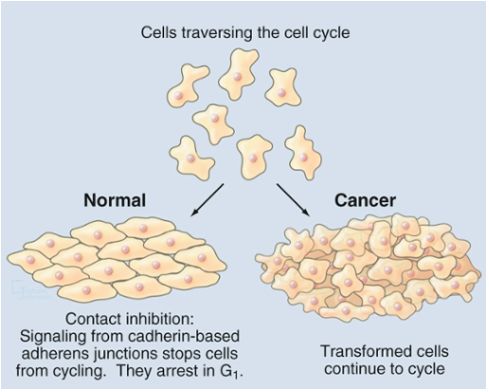
•Checkpoints: controllo delle transizioni
•blocco avanzamento del ciclo fino a quando
la cellula è “pronta”.
Sistema di monitoraggio dei
segnali intra- ed extracellulari
G1 Checkpoint
Check for DNA damage4 checkpoints caratterizzati
G1 early:restriction
crescita cellulare e interazione con la matrice extracellulare
CDK4-cyclin D
G1 late: DNA damage:
integrità DNA prima della replicazione
p53
G2: DNA damage
completamento replicazione
Rad, Hus (sensori)
ATM, ATR, Chk1 (effettori)
Metaphase (spindle assembly checkpoint)
attacco cromosomi al fuso
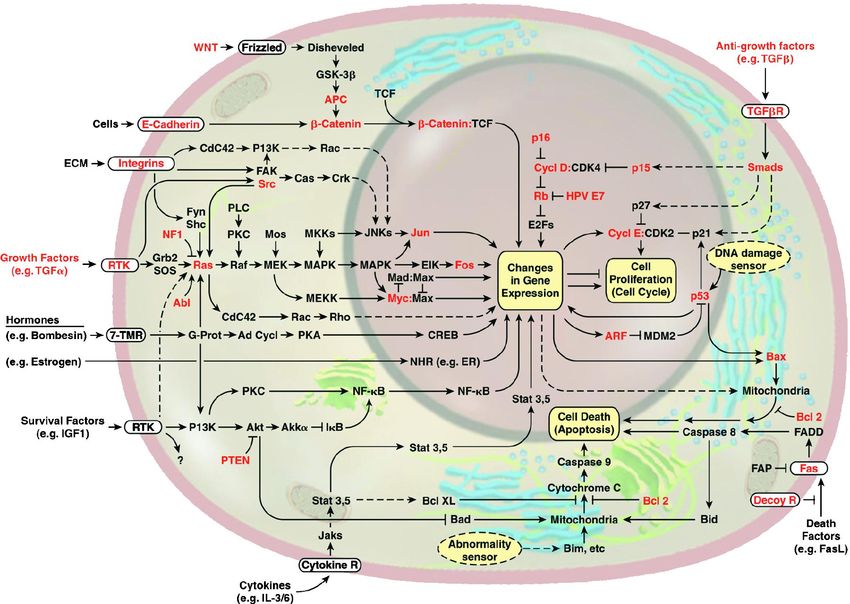
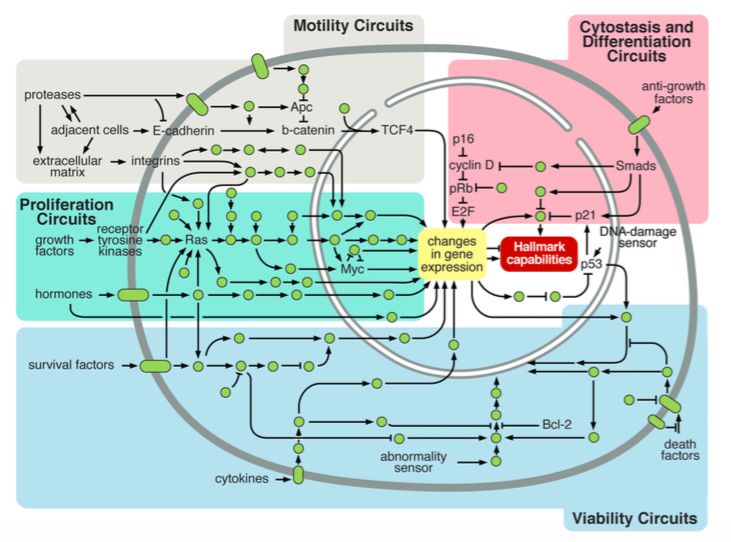
BUB e MADLa cellula come circuito integrato
Due classi di geni che, se mutati, possono
contribuire allo sviluppo tumorale
Oncogeni
virali (v-onc) e cellulari (c-onc)
Se espressi in eccesso promuovono la divisione
20 oncogeni retrovirali (fattori di crescita, trascrizione, chinasi)
Omologhi cellulari con introni, evolutivamente conservati
Attivatori dominanti
Oncosoppressori
Se inattivati non riescono a bloccare la divisione
pRB, p53, pAPC, pBRCA1, pBRCA2Hallmarks of cancer
Processo multistep analogo all’evoluzione darwiniana:
funzioni acquisite che consentono alle cellule
tumorali di sopravvivere, proliferare e diffondersi.
1.
3. 2.
5. 6.
4.G1 Checkpoint Check for DNA damage
Ras wild type
Ras nella cellula tumorale
Mutato nel 25% dei tumori
oncogenec-myc (fattore di trascrizione)
Complessi Myc-Max crescita e replicazione
Complessi Myc-Mad differenziamento terminale
Overespressione di c-myc impedisce il
differenziamento terminale nelle cellule tumorali
Traslocazione reciproca nel linfoma di Burkitt che coinvolge il
gene c-myc (che viene iper-espresso; effetto di posizione)Processes needed for cancer
1.
3. 2.
5. 6.
4.Segnali anti-crescita possono bloccare la proliferazione cellulare in due modi:
1.Passaggio nello stato G0 (eventualmente reversibile)
2.Blocco permanente della proliferazione e inizio differenziamento
G1 Checkpoint
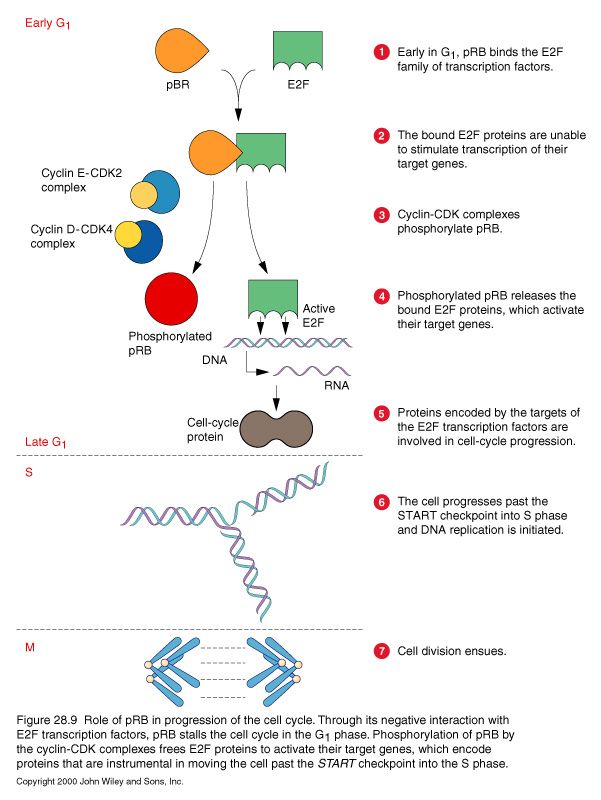
Check for DNA damageSegnali anti-proliferativi convogliati dalla proteina pRb
Retinoblastoma: raro tumore infantile dell’occhio Incidenza 5/100.000 40% mutazione ereditaria 60% sporadico
pRb proteina di 105 kd coinvolta nel controllo del ciclo cellulare Checkpoint G1
• pRb necessaria per mantenere le cellule arrestate in G1
pRb tumorale è inattivo e perde la capacita’ di legare E2F
E2F sempre libero di attivare i geni della divisione cellulare
Proliferazione incontrollata
Rb =oncosoppressoreProcesses needed for cancer
1.
3. 2.
5. 6.
4.3. Evading apoptosis
Espansione popolazione cellule tumorali: proliferazione vs logorio
Apoptosi: morte cellulare programmata
Programma presente in tutte le cellule che, una volta innescato da vari
segnali, si sviluppa in una sequenza quasi “coreografica” di passaggi:
in 30-120 min.
cellular membranes are disrupted,
cytoplasmic and nuclear skeletons broken down,
cyotsol extruded,
chromosomes degraded,
nucleus fragmented
entro 24 hrs il “cadavere” cellulare è “riassorbito”
dalle cellule contigue del tessuto e scompareApoptotic genes
• Apoptotic sensors:
•necessità di mantenere la corretta “architettura” tissutale
-recettori di superficie che legano fattori extracellulari (Fas, TNFalpha
receptors).
-intracellulari (DNA damage, signalling imbalance, survival factor
insufficiency, hypoxia)
• Apoptotic mediators:
Bcl2 family, cytocrome C
• Apoptotic effectors:
Caspases (proteasi)
P53
transcription factor
mutata nel 50% dei tumori umaniG1 Checkpoint Check for DNA damage
Come funziona p53
-P53 regola positivamente Bax
-Bax stimola i mitocondri a rilasciare
Citocromo C e parte il programmaProcesses needed for cancer
1.
3. 2.
5. 6.
4.4. Potenziale replicativo illimitato
Cellule normali: numero limitato di cicli di crescita e divisione
Senescenza Crisi
stato non proliferativo
ma vitale
Acquisizione di potenziale
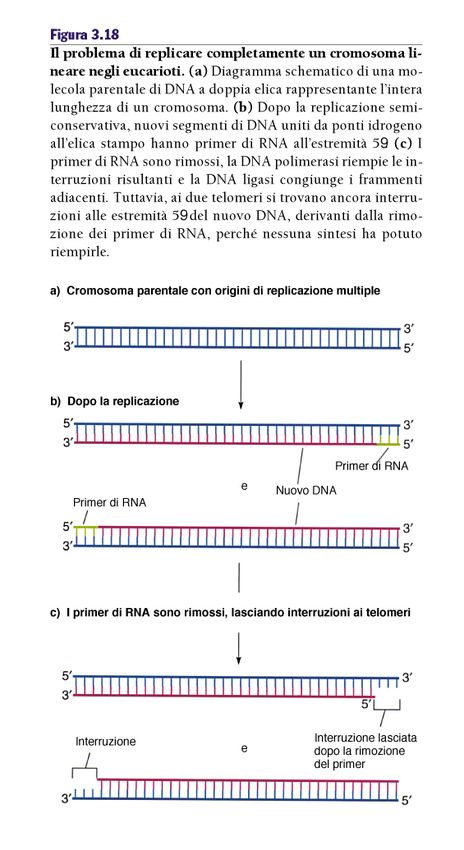
replicativo illimitato morte4. Potenziale replicativo illimitato: Telomeri telomeri: migliaia di ripetizioni di una sequenza di 6bp (TTAGGG) Strutture specializzate che impediscono alle estremità dei cromosomi di frastagliarsi e di fondersi l’una con l’altra, creando uno sbilanciamento dell’informazione genetica Capping telomerico (proteine che si associano alle estremità cromosomiche e le proteggono) Senza telomeri le estremità sarebbero percepite come DNA rotto, che la cellula tenterebbe di riparare determinando un blocco della replicazione e infine morte cellulare
La replicazione
dei telomeri
In assenza di un meccanismo di
compensazione, le estremità dei cromosomi
sono destinate ad essere “corrose” a causa
del meccanismo stesso della replicazione
del DNA, portando (alla lunga) alla perdita
di informazioni genetiche poste agli estremi
dei cromosomi, oltre che alla
“appiccicosità” dei telomeri che a sua volta
può portare alla formazione di aberrazioni
cromosomiche.La telomerasi Tramite un RNA complementare al DNA telomerico, la telomerasi promuove l’allungamento del telomero.
i) Le cellule hanno un potenziale replicativo limitato:
erosione dei telomeri, 50-110bp ad ogni ciclo cellulare
Cellule del sangue: 8.000bp alla nascita, 3.000 mezza età,
1500bp anziani
ii) Una volta esaurito il potenziale (50-70 divisioni) le cellule
si fermano ed entrano in senescenza (G0 irreversibile)
iii) Se p53 e pRb sono mutati le cellule continuano a replicarsi fino
al punto di “crisi” (erosione telomerica, fusioni cromosomiche)
che in genere si conclude con la morte cellulare
1/107 diventano immortali (cellule tumorali)
Up regulation della Telomerasi (85/90%) o del meccanismo ALT
(telomerase independent recombination-based reconstitution of telomeres)Processes needed for cancer
1.
3. 2.
5. 6.
4.5. Sustained angiogenesis
Embryogenesis: new endothelial cells and their assembly into tubes
Vasculogenesis
and sprouting of new vessels from the existing ones
Angiogenesis
Quiescence
Adult: Vasculogenesis transiently turned on (wound healing)
Tumor progression: “angiogenic switch”
that helps expanding neoplastic growth5. Sustained angiogenesis A growing cell must reside within 100µm of a capillary blood vessel •Pro-angiogenic signals Growth factors: Vascular Endothelial Growth Factor (VEGF) up-regulation induced by Ras mutation; Fibroblast Growth factors 1/2) Binding to transmembrane receptors Matrix binders (integrin switch) •Anti-angiogenic signals (normally required to modulate transitory angiogenesis during tissue remodeling and wound healing) Thrombospondin (TSP-1), transcription activated by p53. Endostatin Fragments of plasmin (angiostatin) Angiogenic regulators derived from proteolytic cleavage of Structural proteins
5. Sustained angiogenesis
•Blood vessels produced within tumors are typically aberrant
•Precocious capillary sprouting
•Convoluted vessel branching
•Distorted and enlarged vessels
•Microhemorrhaging
Angiogenic switch from vascular quiescence
early to mid-stage event in human cancer (shared in most common cancers)
Prerequisite for rapid expansion and formation of macroscopic tumorsProcesses needed for cancer
1.
3. 2.
5. 6.
4.6. Tissue invasion and metastasis Invasion and traveling to distant sites, eventual colonization • Local invasion • Intravasation by cancer cells into nearby blood and lymphatic vessels • Transit of cancer cells through the lymphatic and hematogenous systems • Escape from the lumina of vessels into the parenchyma of distant tissues (Extravasation) • Formation of small nodules of cancer cells (micrometastases) • Growth of metastatic lesions into macroscopic tumors • (colonization)
6. Tissue invasion and metastasis Invasion and traveling to distant sites Alteration of proteins involved in tethering of cells to the surroundings: • CAM (cell-to-cell adhesion molecules) N-CAM switch to poorly adhesive forms in neuroblastoma, Wilm’s tumor, small cell lung cancer, pancreatic and colorectal cancer •Cadherins E-cadherin (homotypic cell-cell interactions) lost in most epithelial cancers Integrins (carcinoma switch to alpha3beta1 and alphaVbeta3 subtypes that preferentially bind degraded stromal components)
6. Tissue invasion and metastasis Eventual colonization Metastases: amalgamas of cancer and “conscripted” normal cells extracellular proteases (upregulated) Matrix-degrading proteases produced not by epithelial cancer cells but by conscripted stromal and inflammatory cells
6. Tissue invasion and metastasis
‘‘Epithelial-mesenchymal transition’’ (EMT)
developmental regulatory program, that has become prominently
implicated as a means by which transformed epithelial cells can acquire the
abilities to invade, to resist apoptosis, and to disseminate
A set of pleiotropically acting transcriptional factors
orchestrate the EMT and related migratory processes during
embryogenesis.
These transcriptional regulators are expressed in various combinations in a
number of malignant tumor types.
Causally important for programming invasion in experimental models of
carcinoma formation; some have been found to elicit metastasis when
ectopically overexpressedIntracellular Signaling Networks Regulate the Operations
of the Cancer CellParallel pathways of tumorigenesis
Six capabilities can be acquired in
different orders
A single genetic lesion may confer
multiple capabilities. E.g. p53:
angiogenesis and resistance to apoptosisEnabling characteristics and emerging hallmarks - Switch to “aerobic glycolysis” : glucose metabolism reprogrammed to favor glycolysis to pyruvate versus mitochondrial oxidative phosphorylation - Increase of certain cancers in immunocompromised patients: new evidence that immune system prevents tumor formation and progression also in non virus-induced cancers. Involvement of both acquired and native(NK cells) immune systems
Acquisition of multiple hallmarks largely depending on a
succession of neoplastic cell genomic alterations
Multistep tumor progression as a succession of clonal expansions triggered by
chance aquisition of an enabling mutant phenotype or by epigenetic changes
affecting regulation of gene expression (methylation, histone modifications, etc.)
Cancer cells often increase the rates of mutation
• Breakdown of DNA-maintenance machinery components
(damage sensor and repair genes)
• Compromising the surveillance mechanisms
(mutations in tumor suppressor genes that control senescence and apoptosis)
• Loss of telomeric DNA generating chromosome instability
(double role of telomerase)Enabling characteristics and emerging hallmarks
Tumor-promoting inflammation
Tumors infiltrated by immune cells, mirroring the inflammatory conditions
arising in normal tissues.
Attempt of the immune system to eradicate tumors?
Paradox:
Tumor-associated inflammatory response enhancing tumor progression
helping incipient neoplasia to acquire hallmarks:
• Growth factors sustaining proliferative signaling
• Survival factors limiting cell death
• Proangiogenic factors
• Release of mutagenic chemicals (reactive oxygen species)accelerating
genetic evolution toward malignacyTherapeutic targeting of the hallmarks of cancer
Puoi anche leggere

















































