Infiammazione e tumore - Marco Ciró Dip. Oncologia Sperimentale IEO Via Adamello 16, MI
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
infiammazione e tumore Marco Ciró marco.ciro@ieo.it Dip. Oncologia Sperimentale IEO Via Adamello 16, MI
Infiammazione e cancro
Introduzione
Evidenze di legame infiammazione-cancro
Perché infiammazione aumenta rischio tumore
Via intrinseca/ Via Estrinseca
Oncogeni e infiammazione
Relazione infiammazione cancro: il caso di NF-kB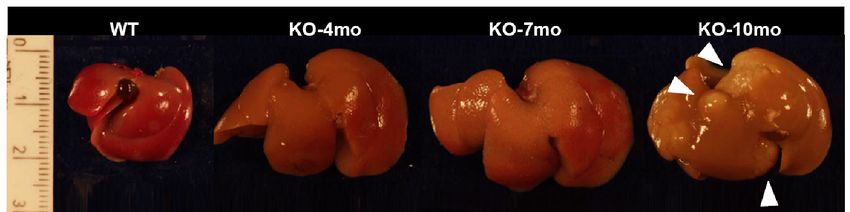
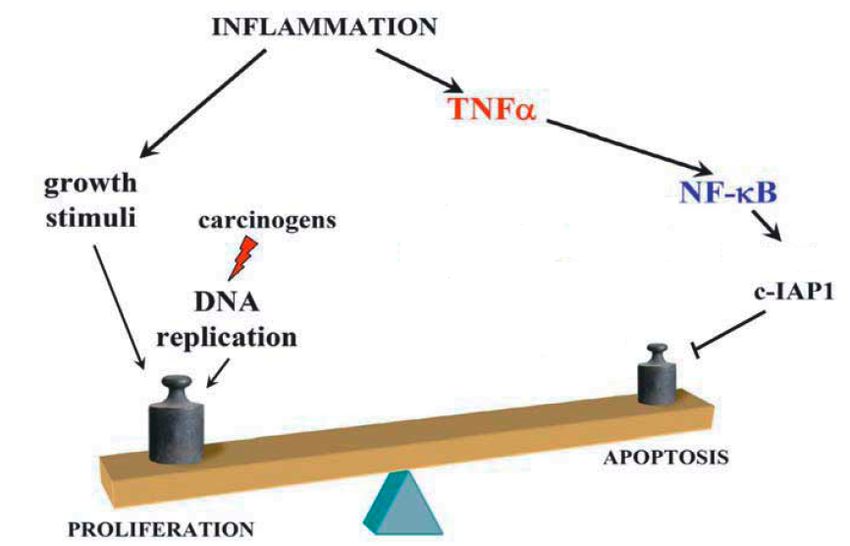
Il cancro é una malattia causata da difetti nei meccanismi cellulari di controllo della stabilitá del genoma e e della proliferazione cellulare Se infiammazione é in grado di stimolare lo sviluppo del cancro, questo deve avvenire attraverso la modulazione di tali meccanismi cellulari Tra i vari pathway attivati durante infiammazione, NF-kB ha un ruolo centrale sia nel controllo della risposta infiammatoria che nello sviluppo del tumore.
NF-kB
NF-kB é un fattore trascrizionale
rapidamente attivato in risposta a stress:
Virus- batteri (esempio: LPS) -parassiti
shear stress (fluido su superficie)
UV-chemoterapia-stress ossidativo
danno al DNA
citochine infiammatorie (esempio: TNF, IL6)
Regola
-attivazione dei linfociti
-infiammazione
-inibisce apoptosi
-stimola crescita cellulare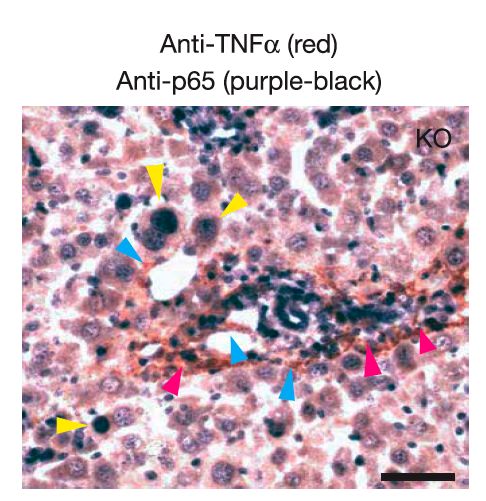
Le subunitá possiedono il dominio REL di legame al DNA REL (la piú importante: REL-A o p65) p50 p52, derivata da proteolisi di p100 Esiste come Etrodimero di due tipi: REL/p50: pathway canonico Regolazione risposta infiammatoria REL/p52: pathway non canonico Organi linfoidi e sviluppo B cell
NF-kB pathway canonico
citoplasma NF-kB inattivo
NF-kB ha segnali di localizzazione nucleare
IkBα lega NF-kB e lo trattiene nel citoplasma
Stimoloè il complesso IkB Chinasi (trimero IKK α,β,γ) inattiva IkBα
NF-kB entra nel nucleo
nucleo NF-kB attivo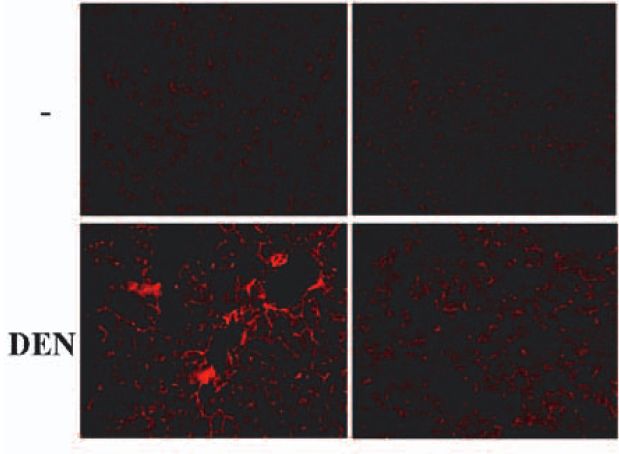
pathway canonico
LPS TNF IL-1
Ionizing
radiation
receptors
Exterior
1
Unknown Cytosol
3 p65 p50
mechanism TAK1
Poly-
I-κB α 4
2b 2a ubiquitin
P P
p65 p50 E3 ligase p65
α p50
γ
β I-κB α α
I-κB Sequestered P P
kinase NF- κB Free NF- κB 5
Proteasomal
p65 p50 degradation
6 of I- κB α
7 Nuclear-
Induces transcription p65 p50 localization
of target genes signals
Nucleus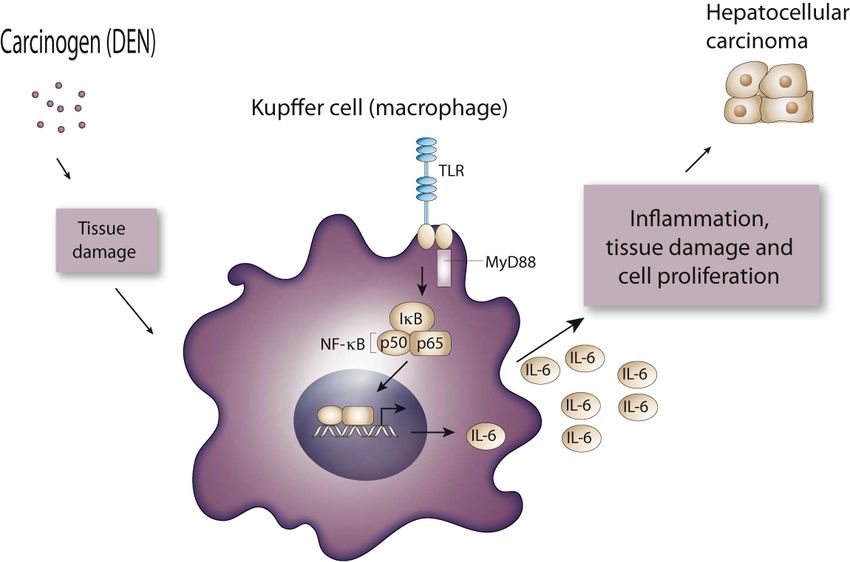
Come inattivare NF-kB? I-kB SR: mutante di IkB che non púo essere inattivato da fosforilazione: = Super Repressore costitutivo
Pathway non canonico
(organogenesi linfoide - sviluppo B cell)
p65 p100
α
α p65 p100
P
IKKα NF-kB
p65 p52
Nuclear-
Induces transcription p65 p52 localization
of target genes signals
Nucleus
Geni target
feedback loop: IkB
proliferazione: MYC, Ciclina D1
anti-apoptosi: BCL2 family (bloccano BAX), cIAP (bloccano caspasi)
Infiammazione:
citochine (e.g., IL-1, IL-6, TNF-a, IFN b, GM-CSF)
chemochine (e.g., IL-8, MIP-1 , MCP1, RANTES, and eotaxin),
molecole per adesione (e.g., ICAM, VCAM, and E-selectin)
effettori, stress ossidativo (e.g., iNOS and COX-2).
risposta immunitaria (MHC proteins)
proteasi (MMP9 proteins)NFKB ha un ruolo sia in cellule infiammatorie che in cellule tumorali
NFKB, infiammazione e cancro Indizi: NF-KB é fondamentale per il processo infiammatorio NF-kB é un attivatore di geni anti apoptotici=oncogeni Tumori umani presentano NF-kB costitutivamente attivato validazione del ruolo di NF-kB in modelli murini Epatite e carcinoma del fegato (HCC:hepatocellular carcinoma) Carcinogenesi chimica e HCC
Hepatocellular Carcinoma (HCC)
High mortality rate due to late stage diagnosis
-resection and transplantation are mostly ineffective
-Commonly used drugs are mostly ineffective
-emergence of therapy resistance tumor
-associated with chronic Liver injury:
HBV infection, HCV infection, alcoholic cirrhosis.
NB HBV increase risk by 100 fold
Prevention!L'epatite virale è un processo infiammatorio (acuto o
cronico), che provoca la morte (necrosi) delle cellule del
fegato (epatociti) a causa dell'attacco da parte di virus
epatotropi (HBV/HCV)
Come risponde l’organo danneggiato?
iperplasia controllatafegato: capacitá rigenerante
iperplasia controllata
Gli epatociti si dividono raramente.
Tuttavia, possiedono la capacità di riprodursi in risposta a:
- fattori di crescita
- distruzione/asportazione di una parte del fegato stesso
Capacitá fondamentale dopo l'asportazione chirurgica o in seguito a lesioni (lesioni virali, tossiche o
ischemiche).
la capacità di rigenerazione del fegato viene
compromessa da danno eccessivo:
- malattie croniche (epatite)
- danni ripetuti (alcol: >50g/giorno/piú anni)
Conseguenza:
epatociti verranno sostituiti da tessuto
cicatriziale (fibrosi) fino a portare alla cirrosi epatica.commento Ripopolazione del fegato e cancro Un ambiente ricco in stimoli proliferativi puó favorire la selezione di una cellula “iniziata” (pre tumorale)
Infiammazione - tumore
Colangite
Infiammazione vie biliari
Hepatocellular carcinomaTopi MDR2-/-
Modello murino di epatite
Malattie epatobiliari:
Gli epatociti secernono la bile nei canalicoli biliari, che sono spazi tra epatociti
adiacenti.
La bile é composta da acidi biliari e fosfolipidi.
MDR2: glicoproteina di membrana che media la secrezione dei fosfolipidi della
bile, famiglia degli ABC transporter.
Topi MDR2 -/- :
Un difetto nella secrezione della bile:
acidi biliari in assenza di fosfolipidi,che sono ritenuti negli epatociti, sono tossici e
causano danno delle membrane e epatite
→modello di malattia epatobiliare, é considerato un modello di malattia cronico
del fegato, precisamente di Colangite, cioé infiammazione delle vie biliari.
All’etá di 4 - 6 mesi i topi mdr2 (-/-) sviluppano foci di HCC.Topi MDR2-/- Modello murino di epatite
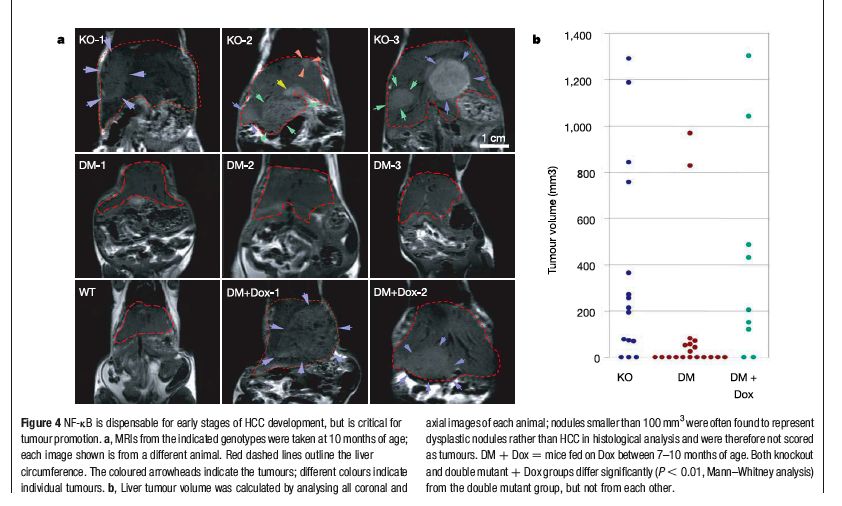
NF-kB e HCC in topi MDR2-/- Sistema: Topi MDR2 -/- sviluppano epatite e poi HCC a 6 mesi Domanda: quale ruolo per NF-kB? Metodo: Inattivazione di NF-kB nel fegato di topi MDR2-/- Come?: Attivazione specifica nel fegato di IkB super-repressor
NF-kB é attivato nei topi
MDR2-/-
+/+ -/- HCC from -/-
Immunoistochimica con antiNF-KB (p65)Cosa succede se inattiviamo NF-kB?
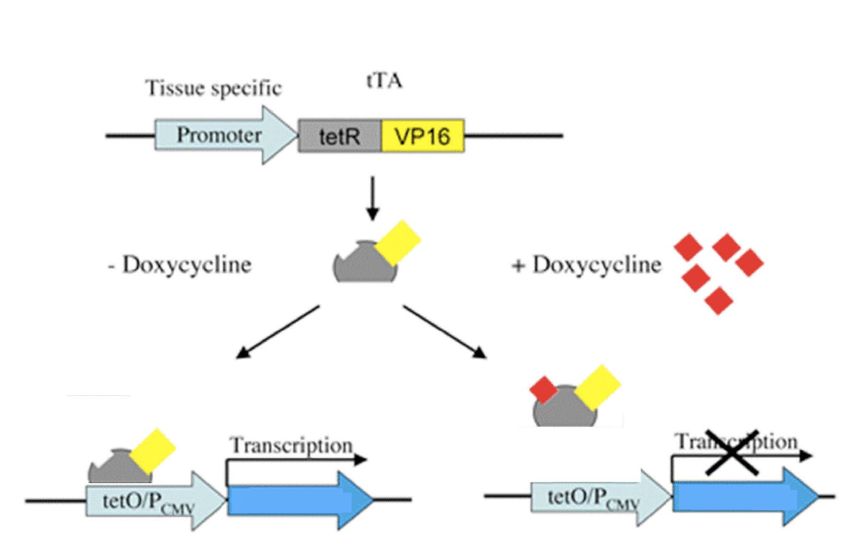
(attivando IkBSR con sistema TET-OFF)
In assenza di DOX, il gene é In presenza di DOX, il gene
acceso, ikBSR inattiva NF-kB é spento, NF-kB é attivo
IkBSR IkBSR
In presenza di DOX, il gene é spento, NF-kB é attivo
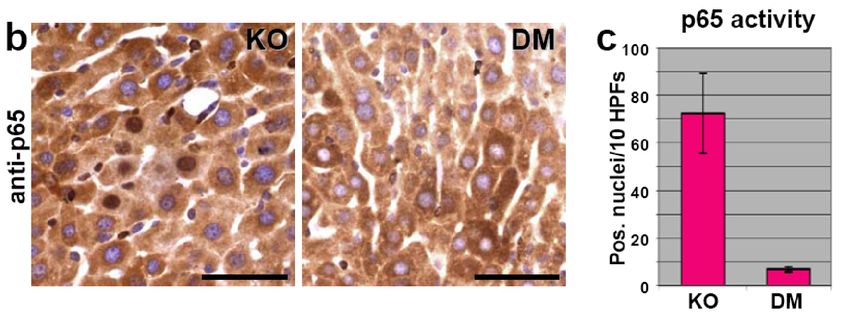
In assenza di DOX, il gene é acceso, ikBSR inattiva NF-kBIkBSR inibisce NF-kB nel fegato Staining nucleare di p65 é indice di attivitá di NF-kB: KO = MDR2-/- DM = MDR2-/-, IkBSR
Se inattiviamo NF-kB? (attivando IkBSR)
meno HCC!
IkbSR spento
IkbSR acceso
MDR2-/-
MDR2-/-
MDR2-/-In che modo NF-kB favorisce
lo sviluppo di HCC
Una funzione di NF-kB é inibire apoptosi.
Ipotesi:
NF-kB favorisce HCC perché inibisce apoptosi
Se cosí fosse, IkBSR dovrebbe far diminuire HCC
perché aumenta il livello di apoptosi.
Quindi:
Qual é il livello di apoptosi in topi MDR2-/- rispetto a
topi MDR2-/- con NF-kB inattivato (da IkBSR)?modello MDR2-/- epatite dox NF-kB IkBSR apoptosi HCC
Inibizione di NF-kB (da parte di IkBSR) aumenta apoptosi e riduce
incidenza HCC
Se blocco TNF, blocco NF-KB: tolgo l’inibizione dell’apoptosi,
e apoptosi aumenta.Quali geni target di Nf-kB? Famiglia IAP: legandosi alle caspasi (anche già attivate) ne inibiscono l’attività enzimatica e ne promuovono ubiquitinazione e degradazione
Quali cellule producono TNF e
quali hanno NF-kB attivato?
NFkB epatociti
endotelio
TNF
macrofagiNFKB funziona sia
in cellule
infiammatorie che in
cellule tumoraliMODELLO di carcinogenesi
chimica di HCC
NF-kB, cellule e citochine infiammatorieCarcinogeni e HCC:
DEN
diethylnitrosamina
DNA alkylation
Induzione HCC in topi adulti:
-Trattamento singolo DEN
-Trattamento con promotore per 6 mesi
-Comparsa HCC intorno ai 6-8 mesi
DEN HCC
promotore
mesiDANNO HCC
DANNO Ruolo NF-kB HCC
DEN
CELLULE NECROTICHE
Attivazione macrofagi
TLR
Produzione di IL6
Stimolazione cell tumorali
HCCTrattamento con DEN induce proliferazione (ripopolamento)
nel fegato
Proliferazione in vivo é dipendente da NF-kB
Wt
NF-kB-/-
Proliferazione misurata nel fegato come % di cellule positive alla BrdUIl trattamento con DEN induce IL6
Questa induzione dipende da NF-kB
WT NF-kB-/-
Immunoistochimica per IL6 su sezioni di fegato da topi wt o NF-kB -/-Funzione di IL6 in HCC indotta da DEN:
In assenza di IL6 si ha meno danno e meno risposta
proliferativa (in vivo)
Danno misurato come rilascio Proliferazione misurata nel fegato
di transaminasi (ALT) come % di cellule positive alla BrdUEsiste un effetto di genere Alti livelli Estrogeni inibiscono rilascio di IL6 e proteggono da HCC
Funzione di IL6 in HCC indotta da DEN: IL6 é richiesta per sviluppo del tumore
E quindi aumenta il survival se togliamo IL6
Quali sono le cellule responsabili del rilascio di IL6?
Kupffer celI (KC): i macrofagi residenti del fegato!
KC isolate e trattate in piastra con LPS o frammenti necrotici:
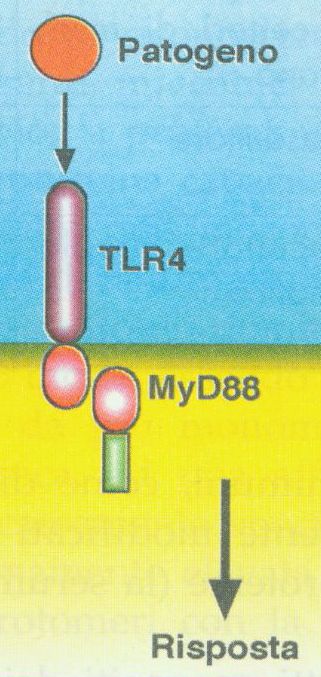
producono IL6 solo se la via di trasduzione del TLR4 é integraNB: naturalmente fonte di “tissue damage” é anche epatite
conclusioni Le cellule e i mediatori dell’infiammazione sono una presenza importante dell’ambiente tumorale Lo stato infiammatorio favorisce insorgenza e progressione del tumore - Infiammazione precede e favorisce l’insorgenza del tumore - Attivazione oncogenica induce un’ambiente infiammatorio che favorisce la tumorigenesi
Terapia anti-infiammatoria come terapia antitumorale Farmaci antiinfiammatori possono avere sia azione profilattica che terapeutica riducono incidenza o rallentano la crescita tumorale: Inibitori delle ciclo-ossigenasi (aspirina, inbitori di COX2) = riduzione incidenza Colon Cancer, Breast cancer Agire sull’infiammazione pro-tumorale: strategie. ¤Inibizione di dei pathway che mediano la crescita/sopravvivenza delle cellule tumorali in risposta alle citochine infiammatorie (inibizione di NFKB) ¤Sequestro delle citochine /chemochine/recettori che mantengono il microambiente infiammatorio (antiIL6, anti TNF, anti CCR2) ¤modulare la risposta infiammatoria che segue la terapia ¤Eliminare cellule infiammatorie responsabili della progressione tumorale, preservando le cellule responsabili della risposta immunitaria ¤Inibizione selettiva di citochine pro-tumorali.
Gli enzimi COX-2 hanno favoriscono di tumorigenesi
COX2 Prostaglandine Blocco apoptosi
AA Migrazione
Neoangiogenesi
Prostaciclina
ALti livelli di COX-2 sono presenti in tumori di diversa origine tissutale:
Breast, Prostate, Pancreas, SKin, LUng, Bladder, Heqad and NEck.
Inibitori di COX-2 sono stati studiati come trattamento anti-tumorale
MECCANISMO:
-COX-2 crea ambiente infiammatorio pro-tumorale
NSAID inibiendo COX-2 bloccano rilascio di metaboliti attivi di Acido Arachidonico.
Controindicazioni: vedi slide precedentiEvidenze epidemiologiche che NSAID riducono rischio tumore 1983 prima osservazione: regressione di polipi al retto in pazienti che assumono NSAID 2012: una serie di tre studi su ampia coorte di pazienti suggerisce uso di Apsirina nel trattamento del tumore e prevenzione di metastasi. utilizzo giornaliero di ASpirina: - riduce il rischio di Colon Cancer -riduce rischio di metastasi. - riduce mortalitá 2015: rischio di effetti collaterali aumentano con etá dei pazienti Trattamento e prevenzione dei tumori Add-ASpirin Trial, 2015 - ongoing " It will recruit 11,000 participants to help find out whether regular aspirin use after treatment for an early stage cancer can prevent the cancer from coming back and preventing death
“therapy induced inflammation”
Ulteriore livello di complessitá
Le principali opzioni terapeutiche è inducono infiammazione
sistemica o locale
Causa: danno tissutale o morte di cellule tumorali
Chirurgia -> danno tissutale /infezioni
Chemioterapia / Radioterapia -> induzione di necrosiPuoi anche leggere

















































