Oncologia (1) Definizioni e caratteristiche generali; criteri di classificazione (2) Basi cellulari e molecolari della cancerogenesi (3) Cause dei ...
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Oncologia (1) Definizioni e caratteristiche generali; criteri di classificazione (2) Basi cellulari e molecolari della cancerogenesi (3) Cause dei tumori (4) Principali caratteristiche cliniche dei tumori
tumore, neoplasia ▪ Denominazione generale, include un ampio gruppo di malattie (ca. 200), caratterizzate dalla
formazione di una massa anomala di tessuto.
tessuto tumorale
▪ il tessuto tumorale è costituito da:
▪ cellule tumorali: cellule con definite alterazioni dei processi di crescita/proliferazione e differenziazione.
▪ stroma: vari tipi di cellule non tumorali, e ECM
▪ La massa del tessuto tumorale si accresce progressivamente, e il processo patologico determina una malattia inizialmente
locale e successivamente sistemica (le cellule tumorali si diffondono e crescono in organi distanti dalla sede di insorgenza).
cellula tumorale
▪ La cellula tumorale (e la sua progenie) è caratterizzata da:
▪ alterazioni nei meccanismi molecolari di controllo della crescita/proliferazione e differenziazione, a loro volta dovute a
più alterazioni genetiche progressivamente acquisite.
▪ nel corso della malattia, le interazioni fra cellule tumorali e stroma si modificano, e si produce un microambiente
favorevole a
▪ crescita del tessuto tumorale (tumore primario)
▪ diffusione a distanza delle cellule tumorali (metastasi).
I tumori: natura del processo patologico • L’organismo è un sistema complesso in cui le singole unità costitutive (>200 tipi diversi di cellule, per un totale > 1014 cellule) sono organizzate in tessuti (insieme di cellule strutturalmente simili e associate dal punto di vista funzionale). • Da una singola cellula uovo fertilizzata a >1014 cellule organizzate in vari tessuti: le cellule staminali embrionarie e le cellule staminali tessuto-specifiche. Tipi di cellule staminali ▪ ESC (cellule staminali embrionarie): nella blastocisti, hanno capacità di rinnovamento virtualmente illimitata, e danno origine a tutte le cellule dell’organismo. ▪ TSC (cellule staminali tessutali o tessuto-specifiche): sono associate alle cellule differenziate che compongono i tessuti, e hanno un repertorio di differenziazione limitato (ad es. dalla cellula staminale ematopoietica deriveranno le cellule del sangue ma non altre cellule).
cosa indirizza il destino di una cellula staminale che si divide verso l’autorinnovamento o la proliferazione/differenziazione?
Modello “stem cell niche”: un compartimento in cui specifici segnali molecolari promuovono l’autorinnovamento delle SC.
Le cellule di Paneth, alla base della cripta, producono il segnale Wnt che opera in modalità iuxtacrina. Quando la SC si divide,
una delle due cellule figlie risulta più lontana dalla cellula di Paneth, fuori dall’effetto di Wnt (quindi fuori dalla nicchia), e
comincia il processo che porterà alla differenziazione; invece la SC figlia che rimane vicino alla cellula di Paneth (alla base
della cripta) continua a ricevere il segnale Wnt e rimane nella sua condizione staminale (autorinnovamento).
In assenza del segnale Wnt, un grande complesso multimolecolare
(destruction complex, DC) assemblato nel citoplasma produce come
effetto la degradazione del co-attivatore trascrizionale beta-catenina.
In presenza di Wnt, il DC è disassemblato, la beta catenina si accumula
e può andare nel nucleo a regolare la trascrizione.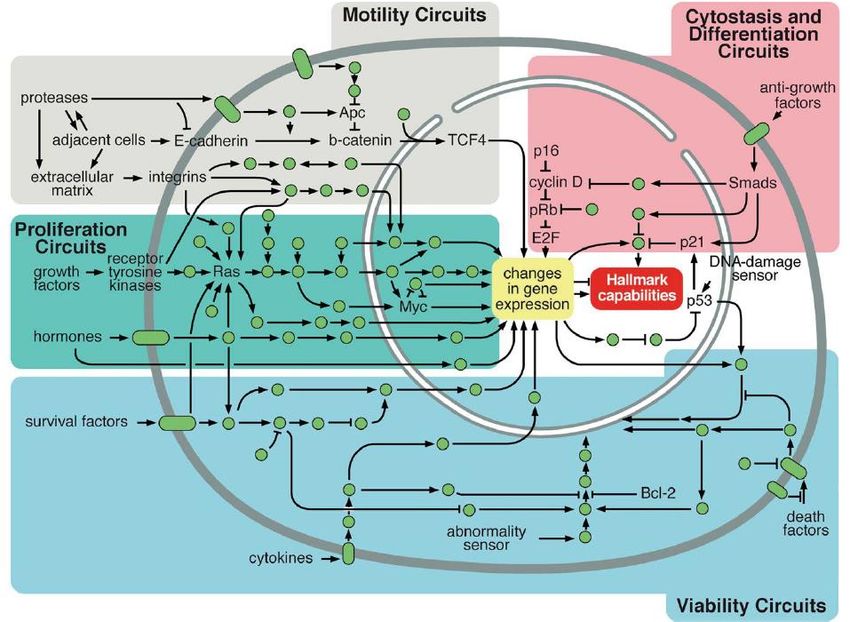
▪ Il modello «stem cell niche» propone il meccanismo alla base del rinnovamento tessutale, e l’apoptosi è il meccanismo di
morte con cui le cellule terminali differenziate sono eliminate dal tessuto.
NB: una cellula staminale nella sua nicchia si divide poco rispetto ad una cellula progenitrice che prolifera perché deve
“popolare” un tessuto di cellule differenziate. Il basso tasso proliferativo protegge la SC dalle mutazioni associate a errori
replicativi e quindi l’integrità genomica di una SC è più garantita. Se poi una mutazione interviene sulle cellule proliferanti,
gli effetti sono contenuti perché queste cellule sono destinate a morire in tempi più o meno rapidi diventando terminali
differenziate.
Le diverse cellule che compongono un tessuto (che si rinnova)
saranno in una condizione di riposo, di crescita, di proliferazione,
di differenziazione oppure potranno aver avviato un processo di
morte. Tali processi sono regolati in modo da garantire il
comportamento «sociale» di ogni cellula, e quindi l’omeostasi e la
funzionalità del tessuto.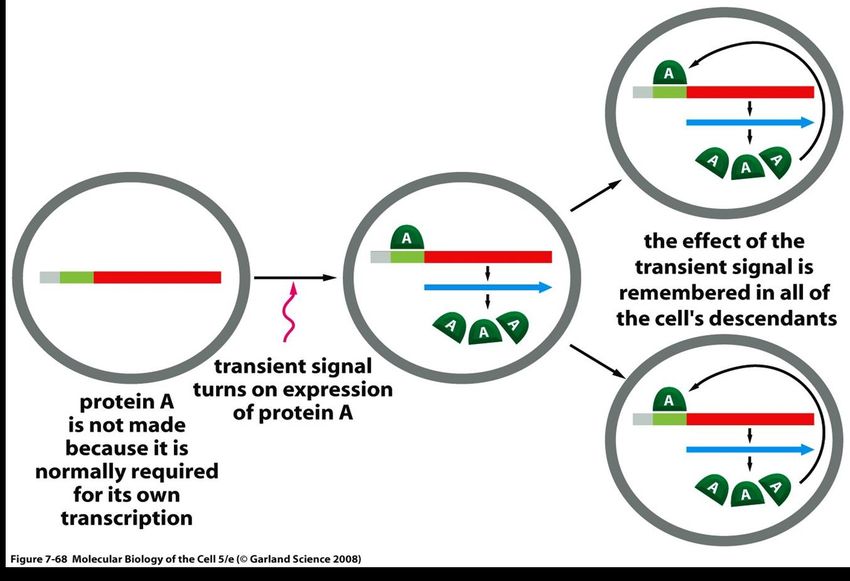
• I tumori sono malattie che originano da una singola cellula appartenente ad un determinato tessuto: in tale cellula, e nella sua progenie, si accumulano nel tempo numerose alterazioni genetiche (genotipo tumorale) che ne modificheranno il comportamento biologico (fenotipo tumorale). ➢ Se le alterazioni genetiche riguardano geni implicati nella regolazione della crescita/proliferazione e della sopravvivenza/morte cellulare, la conseguente eccessiva crescita/proliferazione e il prevalere della sopravvivenza verso la morte produrranno gradualmente una massa anomala di tessuto (tessuto tumorale, tumore primario), a testimonianza dell’alterazione del comportamento sociale della cellula e della sua progenie. ➢ Ogni alterazione genetica che si aggiunge nel tempo sarà assoggettata alla pressione della selezione (sopravvivenza del più adatto). ➢ Quando le cellule tumorali non rispetteranno i confini anatomici del tessuto d’origine (invasione), aumenterà la probabilità che esse si allontanino dal tumore primario e colonizzino altre sedi anatomiche (metastasi), rendendo il processo patologico particolarmente severo.
Principi di classificazione
classificazione clinico-prognostica
▪ tumori benigni: le cellule tumorali hanno acquisito solo alcune delle caratteristiche fenotipiche
tumorali (asportazione della massa → guarigione).
▪ tumori maligni: le cellule tumorali hanno acquisito le caratteristiche fenotipiche tumorali più
aggressive, cioè la capacità di invadere altri tessuti e di metastatizzare (quadro clinico invariabilmente
severo).
classificazione istogenetica
▪ basata sull’identificazione e caratterizzazione morfologica del tessuto in cui le cellule tumorali hanno
avuto origine, e sull’architettura del tessuto tumorale.
ca. 200 distinti istotipi tumorali.
classificazione molecolare
▪ basata sull’identificazione delle alterazioni genetiche responsabili del fenotipo molecolare, si
aggiunge alla classificazione istogenetica (medicina di precisione, o medicina personalizzata).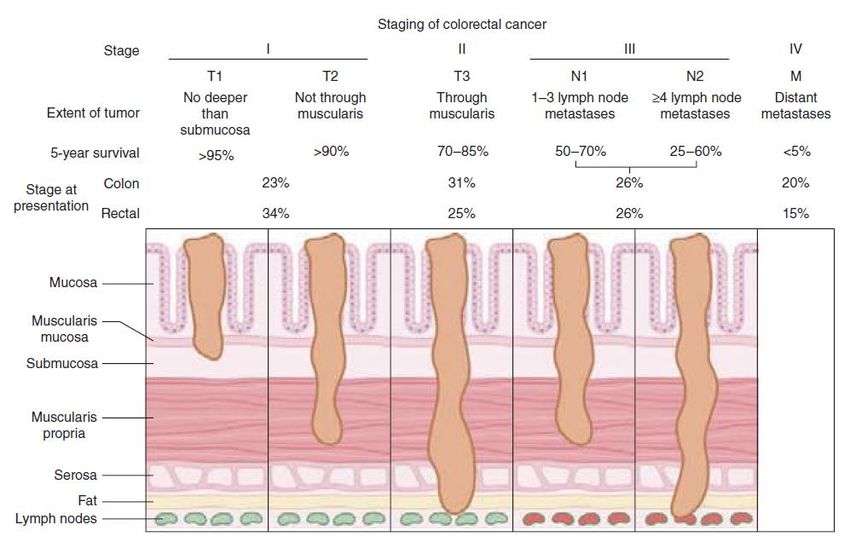
Tumori benigni e tumori maligni: classificazione di significato
prognostico, basata su 4 criteri.
differenziazione tasso di crescita invasione metastasi
cellulare tumorale tumorale
Cellule tumorali ben Crescita localizzata, non
differenziate (uguali Invasiva; confini assenti
benigno Crescita lenta
tumorali ben
alle cellule normali
corrispondenti). demarcati (capsula
fibrosa).
Cellule tumorali con Crescita rapida, con Crescita infiltrativa,
vario grado di possibili variazioni della invasiva (mancato rispetto
maligno differenziazione, da velocità. In genere correla dei confini anatomici del presenti
ben differenziate fino con la differenziazione. tessuto); confini tumorali
all’anaplasia. scarsamente demarcati.
NB: i criteri della differenziazione e del tasso di crescita da soli possono non essere sufficienti a identificare un
tumore come benigno o maligno.
Prognosi: previsione sul decorso/esito di una malattia.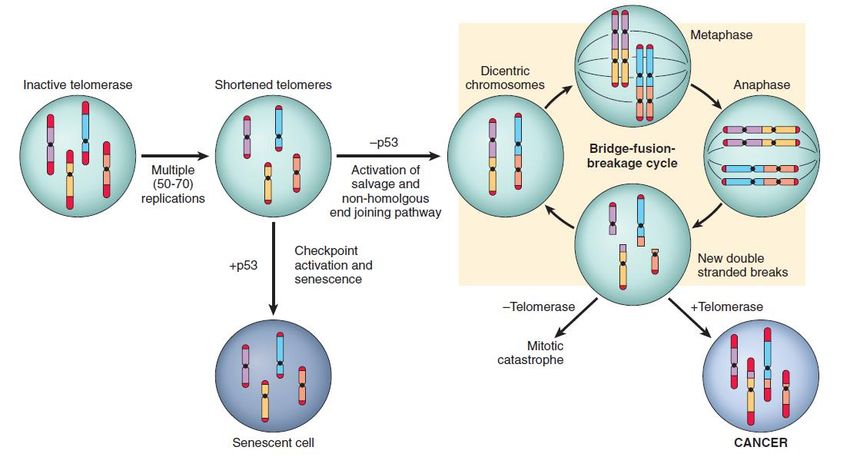
Classificazione dei tumori su base istogenetica
tumori che originano in tessuti di derivazione
mesenchimale
benigni maligni
connettivo e derivati
tessuto fibroso fibroma fibrosarcoma
tessuto adiposo lipoma liposarcoma
tessuto cartilagineo condroma condrosarcoma
tessuto osseo osteoma osteosarcoma
tessuto muscolare
muscolo liscio leiomioma leiomiosarcoma
muscolo striato rabdomioma rabdomiosarcoma
endotelio e correlati
vasi sanguigni emangioma emangiosarcoma
vasi linfatici linfangioma linfangiosarcoma
sinovia - sarcoma sinoviale
meningi meningioma meningioma invasivo
sangue e correlati
cellule ematopoietiche - leucemie
tessuti linfatici - linfomi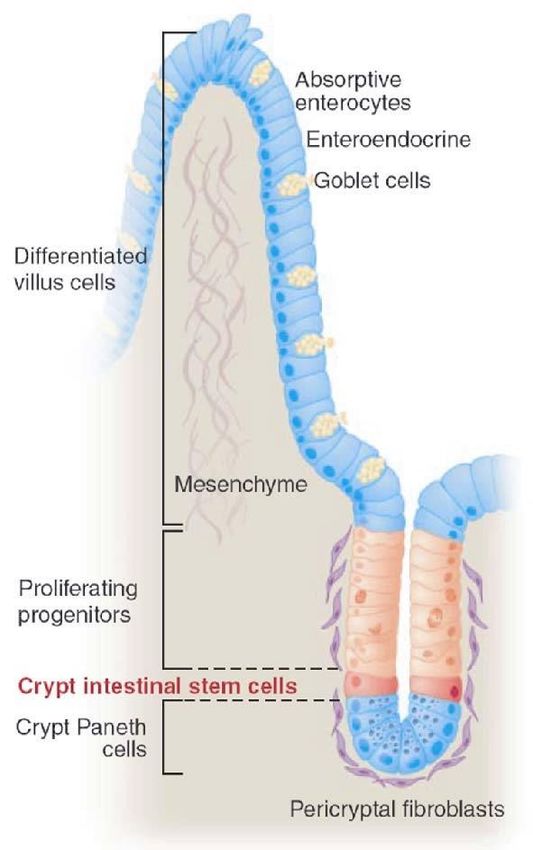
Classificazione dei tumori su base istogenetica
tumori che originano in tessuti di
derivazione epiteliale
benigni maligni
epitelio squamoso stratificato papilloma a cellule squamose carcinoma epidermoide
cellule basali dell’epidermide - carcinoma a cellule basali
adenoma adenocarcinoma
epitelio di rivestimento (ghiandole e
papilloma carcinoma papillare
dotti)
cistoadenoma cistoadenocarcinoma
epitelio di rivestimento albero carcinoma broncogeno
-
respiratorio carcinoide bronchiale
neuroectoderma nevo melanoma
epitelio renale adenoma tubulare renale carcinoma renale
epatociti adenoma epatico epatocarcinoma
epitelio urinario papilloma a cellule di transizione carcinoma a cellule di transizione
mesotelio - mesoteliomabasi cellulari e molecolari della cancerogenesi: principi fondamentali
▪ I tumori sono malattie su base genetica, dovute a più alterazioni mutazionali ed epigenetiche che si accumulano sul DNA
in modo progressivo (cicli successivi di alterazioni, ciascuna seguita da selezione naturale)
• generalmente, le alterazioni mutazionali (cambiamento della sequenza del DNA) sono dovute a esposizione cronica ad
agenti mutageni e all’invecchiamento.
• molto spesso, in aggiunta alle mutazioni, sono presenti alterazioni epigenetiche (cambiamento della modalità di lettura).
▪ I geni in cui sono localizzate le alterazioni responsabili del fenotipo tumorale sono denominati geni tumore-associati, e
possono essere raggruppati in distinte classi funzionali:
• oncogeni: forme alterate, con guadagno di funzione, dei corrispondenti proto-oncogeni che, nella cellula normale,
promuovono la crescita/proliferazione.
• geni oncosoppressori: nella cellula normale inibiscono la crescita/proliferazione, e la loro funzione viene persa nella
cellula tumorale.
• geni implicati nella regolazione dell’apoptosi: geni anti-apoptotici, e geni pro-apoptotici; nella cellula normale, possono
essere considerati rispettivamente proto-oncogeni e oncosoppressori.
• geni implicati nel mantenimento della stabilità genomica (ad es. riparazione del DNA): nella cellula normale, i prodotti
influenzano il tasso mutazionale globale del genoma. [1]
• geni che regolano le interazioni fra cellule tumorali e cellule dello stroma: ad es. cellule dell’immunità, angiogenesi.
▪ Le alterazioni genetiche delle cellule tumorali conferiscono a queste ultime specifiche proprietà biologiche aberranti: le
proprietà biologiche delle cellule tumorali sono denominate collettivamente «fenotipo tumorale».
• La cellula tumorale e la sua progenie acquisiscono progressivamente nel tempo le diverse caratteristiche del fenotipo
tumorale in corrispondenza con l’insorgenza e l’accumulo delle diverse alterazioni genetiche.
[1] Le alterazioni di geni i cui prodotti sono implicati nei sistemi di riparazione del danno del DNA o nel mantenimento della fedeltà
replicativa sono ritenuti di particolare importanza nella progressione della malattia tumorale: infatti un’alterata funzione determina una
condizione di «instabilità genetica» che accelera la velocità di acquisizione delle alterazioni genetiche delle cellule tumorali.La progressione tumorale si attua con cicli successivi di alterazioni genetiche casuali seguite da selezione
naturale.
Modello di acquisizione progressiva delle alterazioni genetiche responsabili del fenotipo tumorale (driver). Si noti che il risultato
finale del processo è rappresentato da una popolazione eterogenea di cellule tumorali (in cui un clone potrà essere prevalente).
NB: un modello del processo di trasformazione di una cellula normale in «cancer founder cell» è riportato nella dia 59.
Esempi di costrizioni ambientali: riduzione della disponibilità di ossigeno e nutrienti; barriere anatomiche naturali che si oppongono all’espansione
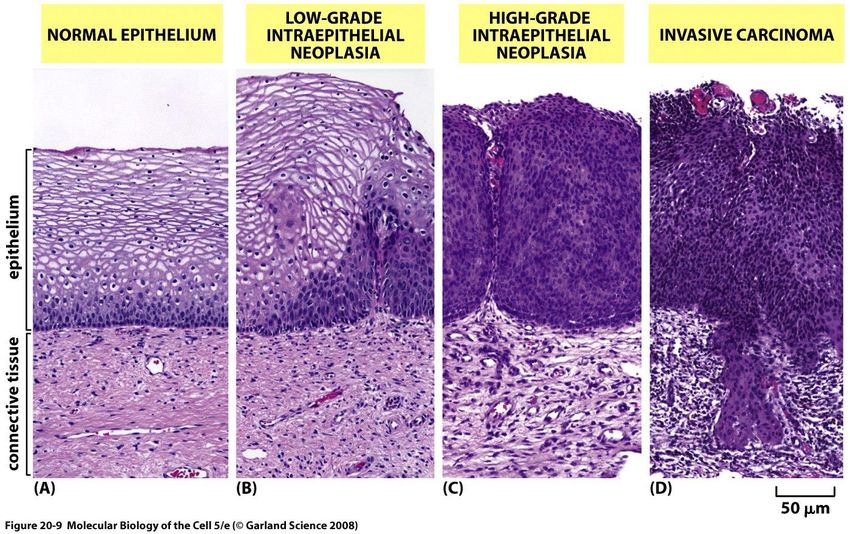
numerica delle cellule tumorali.cervice uterina: progressione tumorale
10% di B 30-40% di CCaratteristiche delle cellule tumorali (fenotipo tumorale)
• capacità di mantenere cronicamente una condizione proliferativa
• capacità di eludere i segnali di soppressione della crescita
• capacità di resistere alla morte cellulare
caratteristiche fondamentali
• capacità di replicarsi in modo illimitato
• capacità di indurre angiogenesi
• capacità di invadere i tessuti adiacenti e metastatizzare in sedi lontane
• capacità di riprogrammare il metabolismo energetico
caratteristiche «emergenti»
• capacità di eludere la risposta immunitaria
caratteristiche «abilitanti» • instabilità genomica
condizioni che favoriscono • infiammazione cronica
l’acquisizione delle caratteristiche
fondamentali ed emergenti.eventi della crescita e proliferazione cellulare
Di norma, fattori di crescita (mitogeni) rilasciati da
cellule stromali sono legati da recettori di
membrana, e da questi parte una cascata di eventi
di segnalazione che regolano la crescita e la
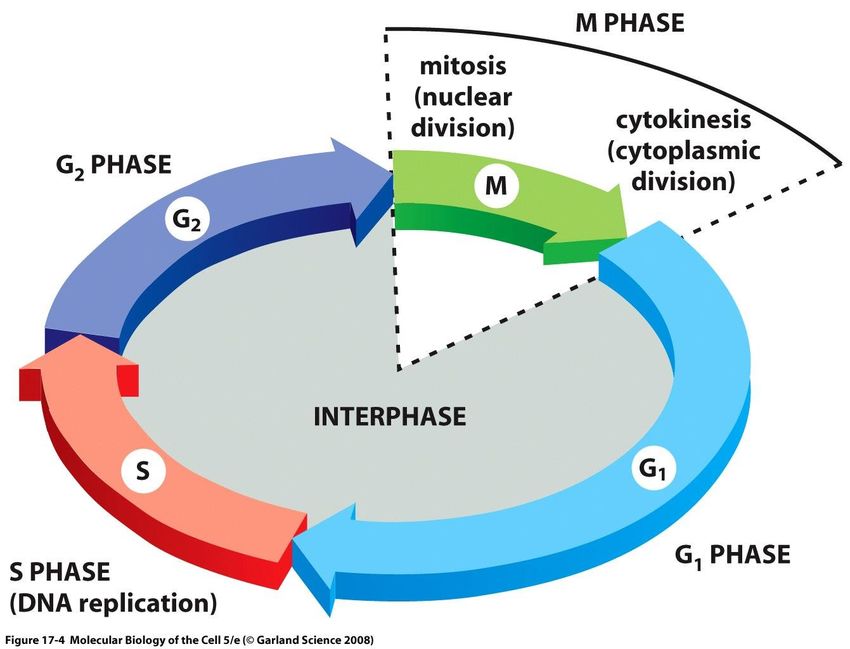
proliferazione cellulare.le fasi del ciclo cellulare
G1 e G2 sono fasi di crescita cellulare e di
monitoraggio dell’ambiente interno ed
esterno. S è la fase di sintesi del DNA, e M è
la fase di divisione.
G1 ha lunghezza variabile, dipendente dalle
condizioni esterne e dai segnali che
vengono dalle altre cellule; qualora le
condizioni esterne non siano favorevoli, la
cellula non progredisce nel ciclo e può
mettersi in una condizione specializzata di
G0 riposo (G0) per un tempo di gg, mm, o
anche anni.
Se arrivano segnali di crescita e divisione, la
cellula in G1 iniziale o in G0 procede in G1
fino al punto start (o punto di restrizione)
start dopo il quale la cellula è commissionata in
S anche se i segnali di crescita sono rimossi.Il ciclo cellulare e il suo controllo
La progressione del ciclo cellulare è
controllata da specifici meccanismi molecolari
(checkpoints) che operano a livello delle
transizioni di fase (G1/S, G2/M,
metafase/anafase).
Su tali meccanismi convergono segnali
molecolari che testimoniano il completamento
dei processi relativi alle specifiche fasi del
ciclo, nonché segnali ambientali.
In presenza di anomalie, la cellula può
interrompere la progressione del ciclo a livello
dei checkpoint, per il tempo necessario a
riparare il danno.cicline, kinasi ciclino-dipendenti (cdk) e controllo della progressione del ciclo
cicline e cdk nel controllo delle fasi del ciclo
fase ciclina CDK
G1 ciclina D CDK4, CDK6
G1-S ciclina E CDK2
S ciclina A CDK2, CDK1
M ciclina B CDK11. capacità di mantenere cronicamente una condizione proliferativa (autosufficienza dei segnali di crescita)
Normalmente, il GFR è attivato transitoriamente dal GF, e
attiva proteine citoplasmatiche che a cascata determinano
la regolazione dell’espressione genica finalizzata a produrre
le proteine necessarie al ciclo cellulare e alla divisione. In
parallelo, vengono espresse le proteine che supportano le
necessarie reazioni metaboliche.
(1) capacità di mantenere una condizione proliferativa cronica,
meccanismi possibili:
• produzione autonoma di GF e GFR (stimolazione autocrina)
• stimolazione della produzione di GF da parte delle cellule
stromali
• aumento dell’espressione di GFR
• modificazione strutturale dei GFR, e avvio del segnale in
modalità ligando-indipendente
• attivazione costitutiva di componenti a valle della via di
trasduzione del segnale *
cause:
alterazioni genetiche a carico di proto-oncogeni, che risultano
“attivati” in oncogeni.
* Si noti che in questo caso non si ricapitola completamente il segnale GF2. capacità di eludere i segnali di inibizione della crescita/proliferazione
segnali di inibizione della crescita • segnali che favoriscono l’uscita in G0
• segnali differenziativi
• segnali di senescenza
• segnali pro-apoptotici
meccanismi di elusione • alterazioni vie RB e p53 (integrazione proliferazione,
differenziazione e apoptosi)
• alterazioni dei meccanismi di inibizione da contatto
• alterazioni della via TGFβ
cause alterazioni genetiche a carico di geni oncosoppressori, la cui normale
funzione risulta compromessa.RB regola il checkpoint G1/S
La perdita del controllo della progressione del
ciclo è una caratteristica biologica
fondamentale delle cellule tumorali.
Per quanto riguarda la funzione RB, almeno
uno dei quattro protagonisti chiave (CDKi,
ciclina D, CDK4, e Rb) è alterato nella maggior
parte dei tumori umani.
Alterazioni di RB nelle cellule tumorali 1. perdita di RB (di entrambi gli alleli) per mutazione
2. alterazioni di geni che controllano la funzione di RB
2.1. iperespressione di ciclina D (per amplificazione o
traslocazione)
2.2. attivazione mutazionale di CDK4
2.3. inattivazione mutazionale di CDKi (inibitori delle CDK)Su p53 convergono segnali da vari sensori di condizioni di stress e anomalie di funzionamento di sistemi intracellulari (ad es. danno al genoma, quantità subottimali di nucleotidi, e/o di segnali che promuovono la crescita, glucosio, ossigeno). Dall’integrazione di tali segnali, p53 può determinare vari effetti, che variano a seconda del tipo cellulare, e dell’entità dell’anomalia (severità e persistenza).
P53 è una proteina inattivata nella maggior parte dei tumori umani
Meccanismo di inattivazione di p53 Conseguenze dell’inattivazione tumori
P53 non riconosce le appropriate
colon, mammella, polmone, SNC,
Mutazione della sequenza DBD* sequenze di DNA e non attiva la
pancreas, stomaco, esofago, e altri.
corrispondente trascrizione
Delezione del dominio carbossi-
Mancata formazione dei tetrameri p53 occasionale, in tumori di varie sedi
terminale
Aumentata stimolazione della
Amplificazione di MDM2 sarcomi, SNC
degradazione di p53
Proteine virali si legano a p53,
Infezione virale inattivandola o favorendone la cervice uterina, fegato, linfomi
degradazione
Incapacità di inibire MDM2 e quindi di mammella, SNC, polmone, e altri casi
Delezione di p14
avviare la risposta p53 con p53wt
Mancanza della funzione di p53 (che
Delocalizzazione di p53 nel citoplasma mammella, neuroblastoma
opera nel nucleo)
*DBD, DNA binding domainSu un’adeguata superficie, le cellule normali
proliferano fino a raggiungere la confluenza, e ciò
determina l’interruzione della proliferazione (inibizione
da contatto).
Al contrario, le cellule tumorali
continuano a proliferare.
Meccanismi molecolari. La proteina merlin accoppia molecole di adesione a recettori di GF,
determinandone il sequestro. In generale, si ritiene che i meccanismi molecolari responsabili
dell’inibizione da contatto sono molti e in larga parte ancora ignoti.La via TGFβ
Il legame di un dimero TGFβ
determina l’assemblaggio di un
recettore tetramerico TGFβR e la
successiva fosforilazione di molecole
Smad. Il complesso trimerico Smad
trasloca nel nucleo e modifica
l’attività di fattori di trascrizione:
▪ attivazione trascrizionale di CDKi
▪ repressione di MYC, CDK2, CDK4,
cicline A ed E), che promuovono la
crescita.
In condizioni normali quindi, il
segnale TGFβ inibisce la
proliferazione cellulare.
In molti tumori, la via TGFβ è alterata a causa di:
-alterazione del recettore TGFβR
-alterazioni di componenti smad
-In molti casi però, pur conservandosi il segnale TGFβ, l’efficacia della via è compromessa da alterazioni
a valle della via TGFβ (ad es. inattivazione di CDKi, iperespressione di MYC).3. capacità di resistere alla morte cellulare
Le cellule tumorali possono eludere l’apoptosi a
seguito di:
(1) Perdita della funzione p53, con conseguente
riduzione di proteine pro-apoptotiche (BAX).
(2) Sovraregolazione di proteine anti-apoptotiche
(BCL2, BCL-XL, e MCL-1) e inibizione di MOMP.
(3) Perdita di APAF-1 (mancata formazione della
piattaforma di attivazione della procaspasi 9).
(4) Sovraregolazione delle proteine IAP (inhibitors
of apoptosis protein).
(5) Ridotta formazione del complesso DISC.
(6) Ridotta espressione del recettore Fas (CD95).4. capacità replicativa illimitata Durante la replicazione, le cellule (telomerasi-negative) accorciano progressivamente i propri telomeri, i quali raggiungono una lunghezza critica in cui gli appropriati checkpoint (G1/S) attivano il programma di senescenza. In assenza di checkpoint, la cellula attiva in modo inappropriato vie di riparazione del DNA (end joining) che portano alla formazione di cromosomi dicentrici. Durante la mitosi, i cromosomi dicentrici sono separati, generando rotture casuali del DNA. Queste ultime, a loro volta attivano vie di riparazione del DNA che associano – sempre casualmente- le estremità del DNA formando ancora cromosomi dicentrici. Più cicli di bridge-fusion-breakage producono un’instabilità cromosomica che ha come esito la “catastrofe mitotica” e la morte cellulare. Tuttavia, se le cellule esprimono la telomerasi possono interrompere il circolo vizioso e sopravvivere mantenendo importanti alterazioni cromosomiche (cellule tumorali).
Il potenziale replicativo illimitato, caratteristico delle cellule tumorali, è una proprietà verosimilmente legata a tre fattori interconnessi: 1. Capacità di eludere il processo di senescenza. Dopo un certo numero di replicazioni, la cellula normale smette di dividersi e comincia ad invecchiare. I meccanismi molecolari coinvolti sono l’attivazione di p53 e di INK4/p16 (forse come risposta all’accumulo di danno sul genoma); l’azione è almeno in parte legata al mantenimento della funzione RB. Nella gran parte dei tumori, il checkpoint G1/S è malfunzionante, e quindi la cellula tumorale eluderebbe così la senescenza. 2. Capacità di eludere la crisi mitotica. Potrebbe conseguire alla riacquisizione della capacità di esprimere la telomerasi oppure al fatto che il processo di cancerogenesi riguarda una cellula staminale (che esprime costitutivamente la telomerasi). 3. Autorinnovamento. Si ritiene che la massa delle cellule che costituiscono un tumore venga alimentata da una «cellula staminale tumorale», che potrebbe derivare dalla trasformazione di una cellula staminale normale, o dalla trasformazione di una cellula progenitrice che riacquista il carattere di staminalità (ovvero di autorinnovarsi). La cellula staminale tumorale ha bassa attività proliferativa, ed è caratterizzata sia dalla capacità di autorinnovarsi sia di produrre cellule progenitrici ad alta attività proliferativa.
vasculogenesis and angiogenesis vasculogenesis angiogenesis Formation of new vessels from Growth of vessel sprouts from existing circulating endothelial cell microvessels. progenitors
angiogenesis pericyte endothelial cell basement membrane
Endothelial cell activation and angiogenesis induction
hypoxia, sex hormones, cytokines, chemokines, growth factors…
tissue cell
tissue cell
VEGF (or bFGF)
receptor protein
relay endothelial cell surface
proteins
gene transcription
cell growth and proliferationAngiogenesis regulation
Epigenetic events Genetic events
Inhibitors Activators
Angiostatin Angiogenin
Endostatin Angiopoietin 1
Estradiol FGF2
IL12 HGF
TIMP IL8
TSP1 PlGF
… VEGF
…6. capacità di indurre e sostenere l’angiogenesi
Il microambiente tumorale può indurre e sostenere l’angiogenesi mediante:
-lo switch angiogenico, cioè modificando l’equilibrio fra induttori (ad es. VEGF-A) e inibitori dell’angiogenesi
(ad es. TSP-1)
-la sovra-regolazione di induttori (come conseguenza dell’ipossia, o dell’attivazione di oncogeni)
-il contributo di cellule di derivazione midollare (cellule della linea mieloide, progenitori vascolari)
la regolazione dell’angiogenesi è una possibile spiegazione……ma molti fattori possono influenzare la «dormienza» del tumore
6. invasione e metastasi
A) Le cellule tumorali si staccano l’una dall’altra, B)producono
enzimi proteolitici capaci di degradare la membrana basale, e C, D)
migrano attraverso lo spazio interstiziale.
Meccanismi patogenetici
-alterazioni dei geni che codificano per molecole di adesione (cellula-cellula, e cellula-ECM)
-sovra-regolazione di geni che codificano per molecole di adesione operative nell’embriogenesi e nell’infiammazione
-attivazione del programma EMT (epithelial-mesenchymal transition): perdita delle giunzioni aderenti e cambiamento
della morfologia cellulare (da poligonale/epiteliale ad affusolato/fibroblastico; espressione di metalloproteinasi della
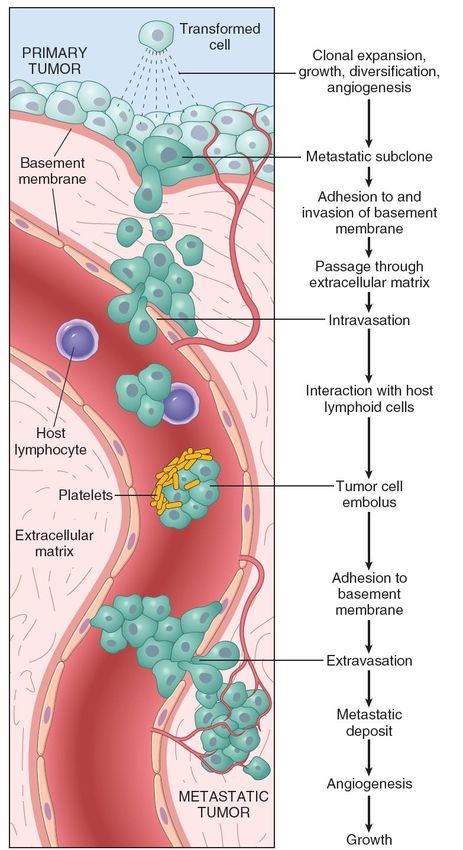
matrice, aumentata motilità, resistenza all’apoptosi.Le varie fasi del processo di metastatizzazione per via ematogena Il processo di metastatizzazione: fasi e ostacoli. Studi sperimentali basati sull’uso di cellule marcate indicano che il processo avviene in fasi distinte, ognuna con un certo livello di difficoltà (rappresentato dal numero di cellule che muoiono nel processo). È nelle fasi indicate che le cellule dei tumori più aggressivi hanno maggiore successo. È opinione generale che la capacità di allontanarsi dal tessuto d’origine e quella di sopravvivere e crescere in una nuova sede siano le proprietà fondamentali del processo di metastatizzazione.
Modelli di metastatizzazione A| metastasi causata da rari cloni varianti che si sviluppano nel tumore primario. B| metastasi causata dalla maggior parte delle cellule del tumore primario caratterizzate da una “metastatic gene signature”. C| combinazione di A e B (cloni varianti in un tumore con metastatic gene signature). D| la formazione di metastasi è influenzata dallo stroma, che regola l’angiogenesi, l’invasione, la resistenza alla risposta immunitaria, e quindi permette alle cellule tumorali di diventare metastatiche.
Caratteristiche abilitanti: Instabilità genomica
Le cellule tumorali accumulano alterazioni genetiche con alta velocità, e i meccanismi molecolari
implicati sono di varia natura (mutazionali ed epigenetici) e tipologia:
▪ Difetti nei sistemi di riparazione del danno sul DNA (DNA mismatch, dsb ed ssb, escissione di
base, escissione di nucleotidi)
▪ Difetti nei sistemi di mantenimento della fedeltà replicativa
Implicazioni dell’instabilità genomica
▪ Da un punto di vista evoluzionistico, l’instabilità genomica è un vantaggio per la cellula tumorale,
direttamente dipendente dall’aumento del numero di varianti e quindi dall’aumentata probabilità di
generare cloni capaci di superare le costrizioni ambientali.
▪ In presenza di un’alterazione strutturale non riparata del DNA, una cellula normale (p53
funzionante) risponderà allo stress interrompendo la replicazione e/o attivando l’apoptosi. Al
contrario, una cellula tumorale con alterazioni p53 potrà sopravvivere (pur in presenza di un
maggiore livello di stress).Caratteristiche abilitanti: infiammazione
• Ad un esame istologico classico, in alcuni tumori sono evidenti numerose cellule dell’immunità innata e
adattativa, così riflettendo una condizione di infiammazione cronica simile a quelle di tessuti non tumorali.
Interpretazione tradizionale: il quadro infiammatorio è legato alla risposta immunitaria finalizzata
all’eliminazione delle cellule tumorali.
• Usando tecniche più sofisticate, si riscontra che virtualmente in tutti i tumori sono presenti cellule
dell’immunità in numero variabile (da poche cellule, visualizzabili solo con Ab specifici, a molte, evidenti
già con colorazioni istochimiche standard).
2000-oggi: sono emersi numerosi indizi che suggeriscono che l’infiammazione può paradossalmente
promuovere la tumorigenesi e la progressione, favorendo l’acquisizione delle «caratteristiche
fondamentali» da parte delle cellule tumorali.
In particolare, le cellule del sistema innato (MØ) possono alimentare il microambiente tumorale con:
• fattori di crescita, capaci di sostenere la proliferazione
• fattori di sopravvivenza, capaci di limitare la morte cellulare
• fattori pro-angiogenici
• enzimi di rimodellamento della matrice (facilitanti angiogenesi, invasione, e metastasi)
• segnali di attivazione EMT
• ROS, attivamente mutageni
Il riscontro che in alcuni casi l’infiammazione sia evidente nelle fasi più iniziali della progressione tumorale,
e sia capace di accelerarne lo sviluppo è a sostegno di un suo ruolo paradossalmente pro-tumorigenico.Caratteristiche emergenti: aapacità di riprogrammazione del metabolismo energetico Il metabolismo energetico di cellule differenziate non proliferanti (sinistra) è basato sulla fosforilazione ossidativa, a meno che una condizione di ipossia non faccia privilegiare la glicolisi, con conversione del piruvato in lattato, allo scopo di rigenerare il NAD+ necessario a sostenere la glicolisi. Le cellule tumorali, e generalmente le cellule proliferanti nel contesto dello sviluppo embrionario o dell’omeostasi tessutale, producono molto lattato anche in presenza di ossigeno, in quanto incrementano la glicolisi aumentando la captazione di glucosio in modo da soddisfare sia la richiesta energetica (ATP) sia i processi anabolici.
Antigeni tumorali riconosciuti da cellule T CD8+
Caratteristiche emergenti: elusione della sorveglianza immunitaria
CARATTERISTICHE BIOLOGICHE DELLE CELLULE TUMORALI E PRINCIPALI MECCANISMI PATOGENETICI
caratteristica biologica meccanismi patogenetici
autosufficienza dei segnali di crescita -produzione autonoma di GF e GFR
-stimolazione della produzione di GF da parte di cellule stromali
-aumentata espressione di GFR
-alterazioni di GFR (attivazione ligando-indipendente)
-attivazione costitutiva di componenti a valle sulla via di trasduzione del senale
insensibilità ai segnali di inibizione della crescita -alterazioni di funzioni che integrano proliferazione, differenziazione e apoptosi
-alterazioni dei meccanismi di inibizione da contatto
-alterazioni della via TGF-β
elusione dell’apoptosi -ridotta espressione di DR
-inattivazione del complesso DISC
-sovraregolazione di componenti bcl2 anti-apoptotici
-sottoregolazione di componenti bcl2 pro-apoptotici della famiglia (spesso perdita di p53)
incremento del potenziale replicativo espressione di telomerasi
difettosa riparazione del DNA instabilità genomica
angiogenesi -switch angiogenico (modificazione dell’equilibrio fra induttori (ad es. VEGF-A) e inibitori
dell’angiogenesi (ad es. TSP-1)
- sovra-regolazione di induttori (come conseguenza dell’ipossia, o dell’attivazione di oncogeni)
- contributo di cellule di derivazione midollare (cellule della linea mieloide, progenitori
vascolari)
invasione e metastatizzazione - alterazioni delle proprietà di adesione (cellula-cellula, e cellula-ECM)
-sovra-regolazione di molecole di adesione operative nell’embriogenesi e nell’infiammazione
-attivazione del programma EMT: perdita delle giunzioni aderenti e cambiamento della
morfologia cellulare (da poligonale/epiteliale ad affusolato/fibroblastico; espressione di
metalloproteinasi della matrice, aumentata motilità, resistenza all’apoptosi.
elusione della sorveglianza immunitaria - incapacità di produrre antigeni tumorali
- alterazioni MHC, alterazioni della processazione dell’antigene
- produzione di citochine immuno-soppressive
GF, GFR (fattori di crescita e corrispondenti recettori). DR (recettori di morte). DISC (death-inducing signaling complex. EMT (epithelial-
mesenchymal transition)Le basi genetiche responsabili del fenotipo tumorale
Genome identity = 99.5% Genome identity = 99%
Le variazioni genomiche interindividuali (che regolano la variabilità della risposta ai fattori ambientali, la
diversa predisposizione alle malattie, le diverse risposte ai farmaci…) sono contenute in < 0.5% del DNA,
circa
15 x 106 bp
forme di variazione di sequenza del DNA nel genoma umano
polimorfismi a singolo nucleotide, SNPs varianti di posizione di un singolo nucleotide; identificati ad oggi
>106 SNPs (1% nelle regioni codificanti, il resto in regioni con
codificanti.
variazioni del numero di copie (CNVs) numerosi e ampi frammenti contigui di DNA sono presenti in
quantità variabili (da 1000 fino a 1.000.000 bp). Il 50% delle CNVs
include sequenze codificanti.Eventi molecolari della cancerogenesi: alterazioni genetiche
▪ Le alterazioni genetiche delle cellule tumorali sono di vario tipo, dalle mutazioni puntiformi
(che interessano singoli nucleotidi) alle anomalie grandi tanto da determinare alterazioni
della struttura dei cromosomi.
▪ In base alle conseguenze funzionali, le mutazioni presenti nelle cellule tumorali possono
essere distinte in:
▪ mutazioni «driver»: interessano i geni tumore-associati e quindi contribuiscono
direttamente allo sviluppo/progressione di un determinato tumore.
▪ mutazioni «passenger»: casuali, diffuse in tutto il genoma, generalmente molto più
numerose delle mutazioni driver. Non hanno un ruolo tumorigenico diretto. Tuttavia, la
presenza di per sé di varianti genetiche conferisce un vantaggio evoluzionistico (ad
esempio, una mutazione passenger potrebbe conferire resistenza ad un farmaco
antitumorale.Alterazioni genetiche nei tumori (principali tipi)
ALTERAZIONE MECCANISMO/I ESEMPI
mutazioni puntiformi ▪ riparazione errata di un’alterazione strutturale del DNA ▪ RAS (ca pancreas, 80%; ca colon, 45%): la
(sostituzione di base, mutazione determina la riduzione
frameshift, etc.) dell’attività GTPasica.
traslocazioni ▪ spostamento di proto-ongogeni dalla normale posizione e ▪ t(8:14); MYC, spostato sul 14 sotto il
iperespressione dovuta al passaggio sotto il controllo di un controllo del promotore del gene delle
promotore forte. catene pesanti delle Ig (linfoma di Burkitt).
▪ produzione di geni di fusione e di proteine chimeriche. ▪ t(14:18); BCL2, spostato sul 18 sotto il
controllo di un promotore di geni per Ig
(linfomi a cellule B).
▪ t(9:22); la fusione BCR-ABL produce una
tirosina kinasi attiva (leucemia mieloide
cronica).
delezioni ▪ perdita di TSG, a cui si aggiungono mutazioni inattivanti ▪ 13q14 (RB)
nell’allele residuo. ▪ 17p (p53)
amplificazioni ▪ duplicazioni di materiale genetico (DNA copy number ▪ MYC (neuroblastoma)
variations) dovute a errori nella duplicazione e/o ▪ ERBB2 (carcinoma mammario)
riparazione, con conseguente iperespressione.
aneuploidia ▪ variazioni del n. di cromosomi, non più un multiplo di 23, per ▪ molto comune nei carcinomi
errori nella segregazione mitotica.In una forma tipica di tumore ci sono 40-100 alterazioni mutazionali che modificano la sequenza delle
proteine, e si pensa che 8-15 di queste sono coinvolte nella cancerogenesi (driver).
Carcinoma colorettale: spettro mutazionale. Sul piano sono
rappresentati da sn a dx i 22 autosomi e il cromosoma X (asse x). La
localizzazione sul cromosoma di ciascun gene mutato è mostrata
dalla posizione del relativo picco (asse y). L’altezza di ogni picco (asse
z) rappresenta la frequenza con cui il gene è mutato in distinti
pazienti. In base all’analisi della frequenza, quelle indicate dai picchi
più alti e intermedi (n=5) sono considerate mutazioni driver nel
carcinoma del colonretto, mentre i picchi più bassi (n=53)
rappresentano mutazioni passenger.
FBXW7: subunità di un complesso ubiquitino-ligasi (target probabile: ciclina E).
PIK3CA: subunità catalitica della PI3K
APC: inibitore della via Wnt; coinvolto anche in adesione/migrazione, attivazione
trascrizionale, e apoptosi.
K-Ras: GTPasi
P53: proteina che risponde a varie tipologie di stress cellulare (vedi)EPIGENETICA: l’insieme delle modificazioni chimiche ereditabili del genoma che modificano
l’espressione dei geni e quindi il comportamento funzionale.
MECCANISMI EPIGENETICI DI REGOLAZIONE DELL’ESPRESSIONE GENICA
I. rimodellamento della cromatina
-Metilazione degli istoni (metil-transferasi, enzimi Specifici enzimi “writers” metilano residui di lisina e arginina, e
demetilanti, proteine che legano DNA metilato) attivano o reprimono la trascrizione in funzione dei residui
metilati.
-Acetilazione (HAT) e deacetilazione (HDAC) degli istoni L’acetilazione di residui di lisina tende ad aprire la cromatina e a
promuovere la trascrizione.
-Fosforilazione degli istoni La fosforilazione di residui di serina può favorire o reprimere la
trascrizione.
-Metilazione del DNA (metil-transferasi, enzimi Alti livelli di metilazione del DNA nelle sequenze di regolazione
demetilanti, proteine che legano DNA metilato) reprimono la trascrizione.
-Fattori di organizzazione della cromatina Proteine che legano regioni non codificanti, coordinano le
relazioni topografiche fra promoter e enhancer.
II. gli RNA non codificanti
-miRNA (microRNA)
-lncRNA (long non coding RNA)modificazioni epigenetiche nelle cellule tumorali
meccanismi epigenetici modificazioni riscontrate nelle cellule tumorali
IPO- E IPERMETILAZIONE GLOBALE IPERMETILAZIONE LOCALE DI PROMOTORI DI TSG
metilazione del DNA espressione genica alterata in più geni inattivazione trascrizionale di TSG
-MODIFICAZIONI ISTONICHE (ad es., ipoacetilazione e ipermetilazione di H3 e H4) sono
frequentemente associate all’ipermetilazione di promotori di TSG.
istoni
-ALTERAZIONI DI ENZIMI DI MODIFICAZIONE DEGLI ISTONI sono state descritte in tumori
ematologici e solidi.
miRNA* i PROFILI DI ESPRESSIONE DEI MIRNA delle cellule tumorali sono diversi da quelli delle cellule
normali, e sono diversi fra differenti tipi di tumore.
* Si noti che i miRNA sono solo un esempio di numerosi tipi di RNA non codificante implicati nel controllo
dell’espressione genica.Il legame fra modificazioni epigenetiche e cancro è chiaro… tuttavia, i meccanismi e le funzioni delle modificazioni epigenetiche (e del loro mantenimento) sono in larga parte sconosciuti, e quindi non è chiaro il significato causale in oncologia. come un circuito di regolazione genica può essere trasmesso alle cellule figlie
Modificazioni epigenetiche e cancerogenesi (iniziazione / progressione) Una modificazione epigenetica può essere una causa “prima” del processo di cancerogenesi, ovvero l’evento iniziante? Quale ruolo possono avere le modificazioni epigenetiche nella progressione? L’idea maggiormente condivisa è che, in relazione al tipo di cancro, il primo evento sia rappresentato da una mutazione (primo hit genetico), seguita da modificazioni epigenetiche (secondo hit epigenetico). In alternativa, modificazioni epigenetiche associate all’età (o ad altro) creano le condizioni per cui gli agenti ambientali siano più dannosi (ruolo iniziante), oppure rilasciano la soppressione della progressione. Per quanto riguarda la progressione, modificazioni epigenetiche possono influenzare i cambiamenti associati all’acquisizione di motilità, invasione, metastasi. Si ritiene che tali eventi, come l’angiogenesi, siano maggiormente controllabili da modificazioni epigenetiche piuttosto che da mutazioni.
Come si classificano gli oncogeni
CLASSIFICAZIONE FUNZIONALE DEGLI ONCOGENI
-fattori di crescita (ad es. PDGF, FGF, IGF-I, HGF)
-recettori per fattori di crescita (ad es. erbB, erbB2)
-proteine di trasduzione del segnale (ad es. abl, ras)
-fattori di trascrizione (ad es. myc)
-regolatori negativi dell’apoptosi (ad es. bcl-2)Come si classificano i geni oncosoppressori
geni che controllano negativamente la proliferazione
molecole di superficie TGFβ-R: gene per recettore di TGFβ, che determina un aumento dell’espressione di p16
con conseguente inibizione di D/cdk.
Caderine: Glicoproteine di adesione intercellulare (cellule epiteliali).
molecole di trasduzione APC (poliposi adenomatosa del colon; la proteina codificata da APC è coinvolta nella
del segnale trasduzione del segnale inibendo beta catenina; senza APC i livelli di beta catenina
aumentano, la proteina trasloca nel nucleo e c’è un aumento della proliferazione).
NF1 (neurofibromatosi tipo 1); è una GTPase activating protein che converte ras da attivo
in inattivo.
molecole di regolazione Rb (vedi), p16 (CDKi)
del ciclo e della
trascrizione
geni che controllano sistemi di riparazione del DNA
Mutazioni ereditarie che diminuiscono la capacità di riparazione del DNA sono associate a rischio oncogeno: XP
(Xeroderma pigmentosum e NER); HNPCC (Hereditary nonpolyposis colorectal cancer, riparo mismatch); BRCA1 e BRCA2
(difetti multipli della riparazione, NHEJ, ricombinazione omologa).
geni che controllano molecole di regolazione dell’apoptosi
p53, e altri geni pro-apoptoticiCaratteristica condivisa di molti tumori è l’alterazione di alcune importanti vie di segnalazione implicate nella sopravvivenza/morte, proliferazione, motilità, senescenza e differenziazione.
Il processo della cancerogenesi (modello) Lo schema illustra un modello di acquisizione graduale di mutazioni «driver», responsabili del fenotipo tumorale, e «passenger», senza conseguenze fenotipiche. ▪ Si noti che, nel modello proposto, l’acquisizione di proprietà staminali (autorinnovamento) e della instabilità genomica siano le alterazioni iniziali che portano alla formazione della «cellula tumorale originaria», da cui poi deriverà con l’acquisizione di mutazioni aggiuntive, sia driver che passenger, una popolazione tumorale eterogenea.
Le cause dei tumori: gli agenti cancerogeni e le loro interazioni cellulari Tradizionalmente, gli agenti cancerogeni sono classificati in tre gruppi: • cancerogeni chimici (suddivisi in «diretti» e «indiretti») • cancerogeni fisici • cancerogeni biologici
Cancerogeni chimici
Cancerogeni chimici diretti Molecole che interagiscono direttamente con il DNA, come gli agenti
alchilanti quali ad es. β-propiolattone, dimetilsolfato, diepossibutano,
ciclofosfamide (e altri farmaci antitumorali), e agenti acetilanti come
l’acetilimidazolo.
Cancerogeni chimici Molecole che interagiscono con il DNA solo dopo attivazione metabolica
indiretti (procancerogeni)
-Idrocarburi aromatici policiclici ed eterociclici (benzoantracene, benzopirene,
dibenzoantracene, metilcolantrene, dimetil benzoantracene)
-Ammine aromatiche, ammidi, azocomposti (naftilammina, benzidina,
acetilamminofluoruene, dimetilamminoazobenzene)
Cancerogeni naturali Aflatossina B1 (da Aspergillus flavus), responsabile di epatocarcinoma in
associazione sinergica con il virus HBV)altri agenti agenti chimici cancerogeni
Asbesto Cloruro di vinile
Cromo, nickel, e altri metalli pesanti Insetticidi (aldrina, dieldrina, clordano)Schema generale degli eventi cellulari e molecolari nel processo di cancerogenesi chimica (cancerogeni indiretti)
Cancerogeni fisici
Radiazioni eccitanti (ultraviolette): responsabili di tumori cutanei
I parametri che influenzano il rischio di sviluppare tumori cutanei sono:
-lunghezza d’onda
-UVA (320-400 nm)
-UVB (280-320 nm)
-UVC (200-280 nm)
-intensità dell’esposizione
-quantità di melanina cutanea
-latitudine
Meccanismo molecolare: Varie alterazioni strutturali delle basi azotate del DNACancerogeni fisici
Radiazioni ionizzanti: radiazioni elettromagnetiche (raggi X e γ) e corpuscolari (particelle α, β,
protoni, neutroni) causano sia tumori del sistema emopoietico sia tumori solidi.
Principali evidenze epidemiologiche
-tumori cutanei nel personale di radiologia (prima delle misure di radioprotezione)
-tumori polmonari in addetti a miniere, in particolare di materiale radioattivo
-leucemie e tumori solidi nei sopravvissuti ai bombardamenti atomici della II guerra mondiale
-leucemie e tumori solidi in esposti a fallout accidentali
Meccanismo molecolare: azione diretta e indiretta (radiolisi dell’acqua) con alterazioni strutturali dello
scheletro zucchero-fosfato del DNA (ssb e dsb).Cancerogeni biologici
Virus a DNA -Papillomavirus (HPV, carcinoma della cervice uterina)
-Herpes virus (virus di Epstein Barr, linfoma; KSHV, sarcoma di Kaposi)
-virus epatite B (HBV, epatocarcinoma)
Meccanismo molecolare:
• il genoma virale si associa stabilmente con il genoma della cellula ospite; nel processo di integrazione
geni virali implicati nel ciclo replicativo vengono interrotti.
• I prodotti di geni virali trascritti nelle fasi iniziali dell’infezione (early genes) interferiscono con i
normali processi cellulari, e favoriscono il processo di cancerogenesi (vedi effetti delle proteine E6 ed
E7 di HPV).
Virus a RNA -HTLV-1 (Human T cell leukaemia virus), endemico in Giappone.
unico retrovirus implicato nella cancerogenesi umana. come HIV, HTLV-1 ha
tropismo per le cellule T CD4+, e l’infezione si trasmette prevalentemente per
via sessuale. Dopo una lunga latenza (10-20 anni), nell’1% degli infetti si sviluppa
una leucemia.
Meccanismo molecolare:
• Il gene virale tax attiva la trascrizione di geni dell’ospite, fra cui vari GFs e GM-CFS che, stimolando la
proliferazione in modo autocrino e paracrino, facilitano il processo di trasformazione tumorale.Effetti trasformanti delle proteine E6 ed E7 prodotte da HPV (HPV16, HPV18)
▪ Capacità replicativa
▪ Proliferazione cronica
▪ Instabilità genomica
Gli effetti complessivi rendono le cellule infette più suscettibili a sviluppare le alterazioni genetiche che conferiscono il
fenotipo tumorale maligno. Nello sviluppo del carcinoma della cervice, hanno importanza patogenetica il fumo, le
coinfezioni microbiche, le modificazioni ormonali, e le deficienze dietetiche.Caratteristiche cliniche dei tumori
caratteristiche cliniche dei tumori (benigni e maligni)
localizzazione -compressione e distruzione di strutture adiacenti
-ostruzione (ad es. intestino)
attività funzionale -produzione di ormoni (tumori benigni del sistema endocrino)
-produzione di ormoni e sostanze ormono-simili (tumori maligni non
endocrini)
ulcerazioni, infezioni facilmente determinate dalla crescita invasiva e distruttiva della massa
secondarie, sanguinamento tumorale
cachessia tumorale -calo ponderale progressivo (massa muscolare e grasso)
-anoressia
-anemia
-astenia
-segni sistemici dell’infiammazione (ad es. proteine della fase acuta)
sindromi paraneoplastiche segni e sintomi (endocrini, neuromuscolari, dermatologici, osteo-articolari,
vascolari) non facilmente riferibili al tumore in base alla sede anatomica e
all’istotipo.
dolore variamente presente in tutte le condizioni tumoraliInquadramento clinico del paziente: grading e stadiazione
grading (grado): valutazione semi-quantitativa della differenziazione di un tumore maligno.
• Basato sul grado di differenziazione delle cellule tumorali e sul numero di mitosi.
• Espresso come grado I-IV, in correlazione alla crescente anaplasia.
• Ogni forma di neoplasia ha parametri di grading specifici, ma in generale si giudica
quanto una cellula tumorale assomigli (o non assomigli) alla controparte normale.stadiazione: valutazione della diffusione locale e sistemica della malattia.
• I criteri utilizzati si basano sulle dimensioni della lesione primaria, sulla diffusione verso i linfonodi
regionali, e sulla presenza o meno di metastasi ematogene.
• TNM (metodologia più diffusa)
• T, dimensioni del tumore primario; T0 = in situ; T1-T4 (dimensioni crescenti).
• N, linfonodi; N0 = nessun linfonodo; N1-N3 = coinvolgimento crescente di linfonodi e stazioni
linfonodali.
• M, metastasi; M0 = no metastasi ematogene; M1 ( a volte M2) metastasi ematogene (con eventuali
giudizi sul numero).
NB: La stadiazione è al momento la migliore e più affidabile premessa per la scelta della miglior forma di
terapia.
«L’aggressività» di un tumore dipende dalla variabile combinazione di tre caratteristiche clinico-biologiche: alta capacità proliferativa delle
cellule tumorali, alto potenziale metastatico delle cellule tumorali, e resistenza alla terapia.Carcinoma del colon-retto: stadiazione e implicazioni prognostiche
La prevenzione dei tumori • Prevenzione primaria: individuazione (ed eliminazione) delle cause; basata su indagini epidemiologiche (registri di morbilità e mortalità) e di laboratorio (test di mutagenesi, test di cancerogenesi). • Prevenzione secondaria: diagnosi precoce, ovvero diagnosi in assenza di sintomi (o con sintomi e segni generici); basata su indagini epidemiologiche, diagnosi di laboratorio, e procedure medico-chirurgiche. Prevenzione secondaria di tumori dell’apparato genitale femminile (mammella, cervice), di tumori gastrointestinali (anemia ipocromica). • Prevenzione terziaria: prevenzione e/o individuazione tempestiva della ripresa di malattia; basata su procedure medico-chirurgiche).
Puoi anche leggere

















































