IL DOMINIO N-TERMINALE DELLA PROTEINA PRIONICA: CARATTERISTICHE E MECCANISMI PATOGENETICI - 3 GIUGNO 2015
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
IL DOMINIO N-TERMINALE DELLA
PROTEINA PRIONICA: CARATTERISTICHE
E MECCANISMI PATOGENETICI
3 GIUGNO 2015
Dott. ssa Livia Bernardi
Biologo Genetista
Laboratorio di Genetica Formale e Molecolare
Centro Regionale di Neurogenetica, ASP CZ
GENE DELLA
PROTEINA PRIONICA
(PRNP)
GENOTIPI…..
MUTAZIONI
DEL GENE
PRNP
Le MUTAZIONI a livello del gene PRNP possono essere di 3 tipi:
• mutazioni puntiformi → mutazione “missense” →sostituzione di un AA con un altro
• mutazioni puntiformi → mutazione “nonsenso ” → formazione di codone di stop originariamente non
esistente
• inserzioni o delezioni di octapeptidi extra (nella regione N-terminale sono normali 5 repeats di
octapeptidi fra gli aminoacidi 51 e 91: la presenza di più di 3 octapeptidi extra è correlata a malattia
da prioni ereditabile).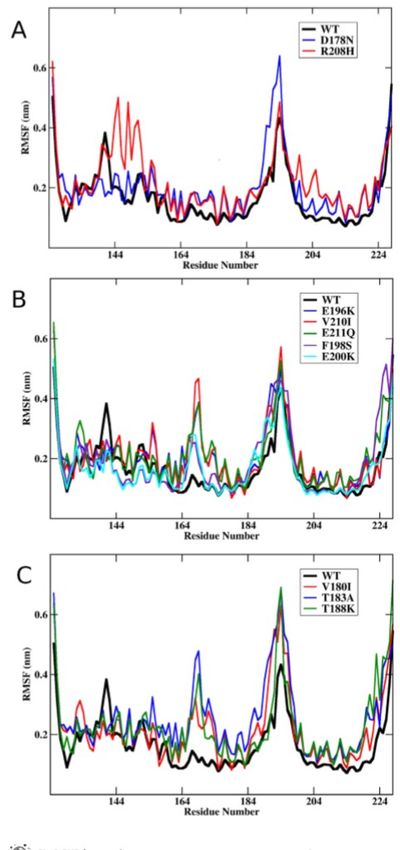
Mutazione: frequenza ≤ 1 su 100
Polimorfismo: frequenza > 1 su 100
Mead S., Eur J Hum Genet. 2006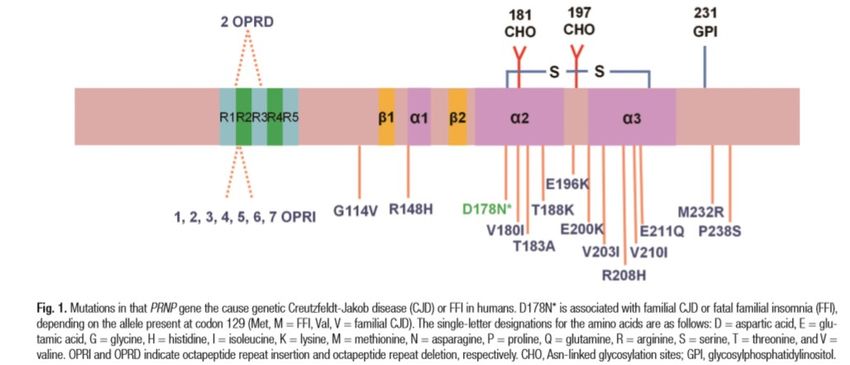
…….E FENOTIPI

VARIABILITA’ FENOTIPICA
ASSOCIATA A MUTAZIONI NEL
GENE PRNP
• Sono state scoperte più di 30 mutazioni diverse a carico del gene PRNP,
tutte responsabili di forme di Malattia da prioni;
• Ampio spettro di variabilità fenotipica sia per quanto riguarda la
sintomatologia clinica, (GSS, Gerstmann-Straussler-Scheinker, FFI, Familial
Fatal Insomnia e CJD, Malattia di Creutzfeldt-Jacob), sia per quanto
riguarda l’età di esordio e la durata della malattia, anche nell’ambito di una
stessa famiglia, in parte spiegabile con il polimorfismo PRNP M129V.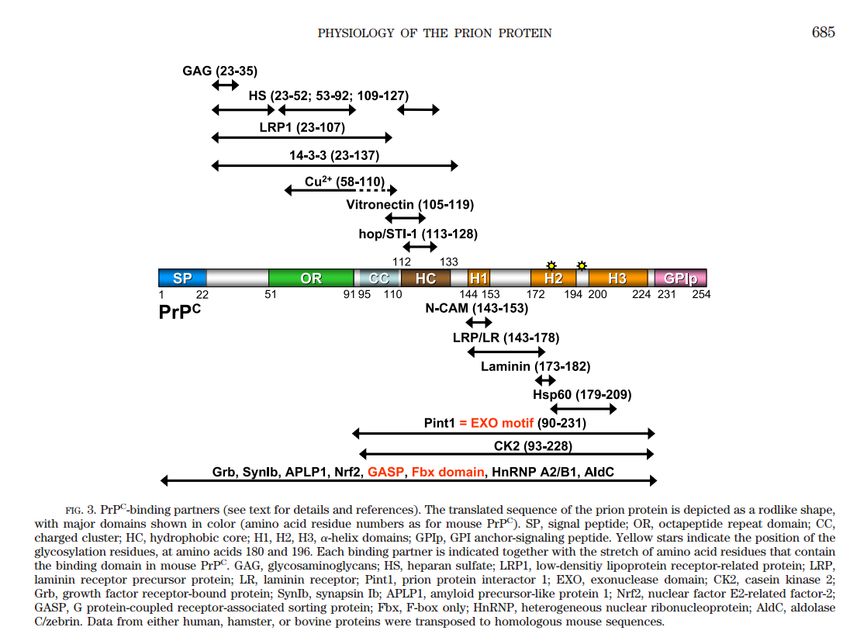
Asp
218 Tyr→Asn
(Alzualde et al., 2010)
Jeong BH et al., JKMS 2014

MUTAZIONI NEL GENE PRNP RIPORTATE IN ASSOCIAZIONE A
FENOTIPI ATIPICI FTD-LIKE…..
• Nitrini et al., Arq Neuropsiquiatr 2001: mutazione Thr183Ala in una grande famiglia (12 affetti) con
Malattia da prioni accertata con neuropatologia con fenotipo clinico assolutamente simile alla
Demenza frontotemporale con parkinsonismo.
• Hall et al., Neurology 2005: mutazione H187R in una famiglia con demenza frontotemporale,
trasmessa con eredità autosomico dominante, anticipata nell’adolescenza da disturbi psichiatrici. La
malattia è caratterizzata da lunga durata. La neuropatologia mostra atrofia e modesta gliosi.
• Woulfe et al., 2005: mutazione Q217R in una paziente esordita a 45 anni rivelatasi una GSS con
fenotipo clinico somigliante a demenza frontotemporale (diagnosticata inizialmente come Malattia di
Pick). Neuropatologia consistente con GSS.
• Clerici et al., J Neurol 2008: mutazione Glu196Lys in una paziente di 75 anni senza storia familiare
per demenza con originariamente diagnosi di Demenza frontotemporale.
• Giovagnoli et al., Neurol Sci 2008: mutazione Pro102Leu in una famiglia con fenotipo di demenza
frontotemporale, qui riportato come nuova espressione fenotipica della malattia di Gerstmann-
Straussler-Scheinker.
• Kumar et al., 2012: inserzione di 12 octarepeat (12 OPRI) in una famiglia con FTD atipica e
atassia. Neuropatologia consistente con malattia da prioni (placche PrP positive e diffuse tangles tau-
positive).
…..E ALTRI FENOTIPI ATIPICI
Codon Amino acid change cis-codon 129 Reference
PrP Angiopatia Cerebrale Amiloide
(CAA) (mutazione nonsenso)
145 tyrosine to stop methionine Ghetti 1996
(Kitamoto 1993b)
Codon Amino acid change Phenotype Reference
171 Asparagine to serine(cis Familial neuro- psychiatric Samaia 1997
valine at codon 129) illness (psychotic
Altri fenotipi
depression, violent (mutazioni
behaviour, some also have missense)
dementia; others only
psychiatric problems).
183 Threonine to alanine Familial Alzheimer’s Nitsch 1998
disease; 4 year duration
(with dementia)
6 octapeptide repeat (144 bp) insert Variable including CJD, GSS, "Alzheimer’s", atypical Collinge
dementia, "Huntington’s", etc. 1992
Altri fenotipi
9 octapeptide repeat (216 bp) insert Owen 1991
sporadic dementia; 2.5 year duration; no
spongiform change but PrP+ve plaques.
(OPRD, inserzioni
di octapeptidi)LA PROTEINA
PRIONICA
• Glicoproteina di circa 250 amino acidi.
• Altamente conservata nei mammiferi (regione fra i residui 23-90
(Wopfner et al., 1999; Kim et al., 2008).
• Espressa abbondantemente nei neuroni (principalmente nelle
terminazioni sinaptiche), ma anche nel sistema linforeticolare e nei
muscoli scheletrico e cardiaco (Horiuchi et al., 1995).
• Si localizza sulla superficie esterna della membrana plasmatica
attraverso un’ancora di glicosil-fosfatidil-inositolo (GPI), in siti specifici
ricchi in colesterolo e glicosfingolipidi, denominati rafts (Taylor et al.,
2006).
• Tuttavia, alcuni dati evidenziano come essa possa risiedere anche in altri
distretti della membrana ricchi di clatrina, dai quali originerebbe il
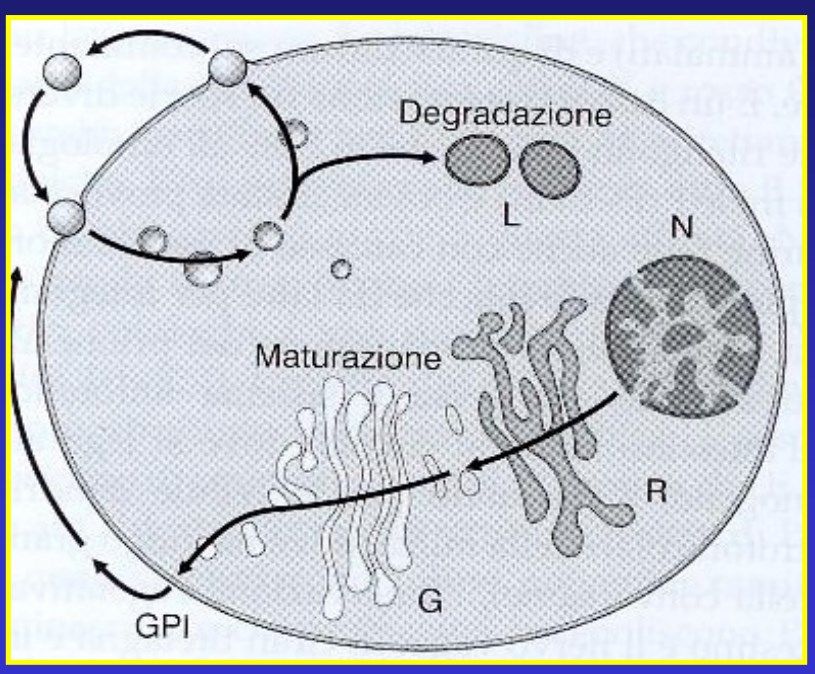
processo endocitotico della proteina (Magalhaes et al., 2002).CICLO DELLA PROTEINA PRIONICA
• E’ proteina di secrezione →viene sintetizzata nel
La PrPC si muove
reticolo endoplasmico (R) per poi essere portata, dopo
ciclicamente dalla membrana essere transitata nel Golgi (G), alla membrana
plasmatica al compartimento plasmatica (Orsi and Sitia, 2007).
endosomiale e viceversa. • Durante questo tragitto, va incontro a maturazione
attraverso modifiche post-traduzionali. La prima è il taglio
del peptide segnale (i primi 22 amino acidi dell’N-
terminale, dirigono la proteina al reticolo endopalsmico),
seguita dall’attacco dell’ancora GPI al C-terminale (al
residuo Ser 231 sempre nella sequenza umana) e dalla
formazione del ponte disolfuro tra i due residui di
cisteina 179-214 (nell’uomo). Altrettanto caratterizzanti
sono le aggiunte di zuccheri complessi nei residui di Asn
181 e 197, e ciò comporta che la PrPC possa esistere in
isoforme diverse aventi una, due, o nessuna, ramificazione
glucidica.
• Dopo essere stata internalizzata, la proteina prionica viene
riciclata dopo essere stata direzionata ai lisosomi (L) per
essere degradata.FUNZIONI
DELLA PROTEINA
PRIONICA
• Ruolo nel metabolismo del rame (Brown et al., 1997), e, dato che lo ione è parte essenziale di enzimi
coinvolti nella rimozione delle specie reattive dell’ossigeno (ROS), anche nella protezione contro lo
stress ossidativo, dato che questa lega selettivamente Cu2+ per trasportarlo attraverso la
membrana plasmatica (Cu2+ stesso stimola l’endocitosi della proteina (Pauly et al., 1998); la rimozione
dalla PrPC del dominio degli octarepeats, o la mutazione delle istidine che lo legano, aboliscono
l’effetto.
• Adesione, migrazione e differenziamento cellulare, mediante l’interazione con diversi putativi
partner funzionali (ligandi, altre proteine ma anche lipidi, acidi nucleici e metalli) che includono
molecole di adesione cellulare, proteine di membrana e della matrice extracellulare (Sorgato and
Bertoli, 2009). Tali interazioni medierebbero eventi di trasduzione del segnale, che, in linea con la
generale proprietà di cito-protezione della proteina, contrasterebbero specialmente la morte
apoptotica della cellula (rispetto ai WT, i neuroni di topi PrP-KO si sono maggiormente suscettibili
all’apoptosi indotta da deprivazione di siero (Kuwahara et al., 1999).LIGANDI DELLA PROTEINA PRIONICA
Linden et al., 2007STRUTTURA DELLA PROTEINA PRIONICA
• La proteina matura è divisa in due domini: dominio N-terminale (residui 23-120) e dominio C-terminale (residui 121-
231), strutturalmente indipendenti (Riek et al., 1996).
• Tutte le mutazioni patogene finora identificate nel gene PRNP si trovano nel dominio C-terminale o nel dominio N-
terminale a partire dal codone 102, con ECCEZIONE delle OPRD e OPRI (residui 51-91, che formano un segmento di
cinque copie in tandem di una sequenza di otto aminoacidi (octarepeat), e della mutazione Pro39Leu (Bernardi, Cupidi,
Frangipane et al., Neurobiol aging 2014), che invece sono localizzate nel dominio N-terminale.
• Questo ha portato a spostare tutta l’attenzione dei ricercatori sulla regione C-terminale (regione amiloidogenica, molto
conservata nei mammiferi) che è ritenuta associata alla malattia, per cui è stata più studiata della regione N-terminale
(Nunziante et al., 2003).Gill AC et al., EMBO J 2000
• La regione N-terminale è ampiamente non strutturata e flessibile , e ciò le dà il vantaggio della possibilità di
interagire con diversi ligandi, micro-(ioni rame) e macro-molecole (lipidi, proteine), a differenza delle strutture
stericamente rigide; comprende un segmento di cinque copie in tandem di una sequenza di otto aminoacidi
(octarepeat), implicato nel legame col rame, in assenza del quale non assume struttura secondaria (Brown et al., 1997).
Ha un alto grado di conservazione nelle specie (Wopfner et al., 1999), in particolare i residui 23-90, il che suggerisce
un loro importante significato funzionale (Wopfner et al., 1997; Kim et al., 2008).
• La regione C-terminale forma invece un rigido dominio globulare, contenente 3 regioni ad alfa-elica e due a
foglietto beta antiparalleli. Termina con un’»ancora» di GPI (glicosil-fosfatidilinositolo) che lega la proteina alla
superficie esterna della membrana cellulare. Questo dominio è stabilizzato da un ponte disolfuro che collega l’elica α2
con l’elica α3 e comprende due siti di glicosilazione (Donne et al., 1997).PROTEINE NON STRUTTURATE FLESSIBILI
(DOMINIO N-TERMINALE)
• Proteine la cui funzione è direttamente
correlata al disordine strutturale
Trasduzione del segnale
• Hanno una conformazione estesa
(quindi riconoscono molti ligandi) che Regolazione del ciclo cellulare
dipende dalla
Espressione genica
• Composizione AA caratteristica con
abbondanza di prolina, non casuale.
TRANSIZIONE DISORDINE-ORDINE: può consistere sia nell’assunzione di uno stato
semplicemente più ordinato, sia di una struttura secondaria o terziaria.
BINDING PROMISCUITY: capacità di legare più target differenti. Ovviamente ciò
presuppone l’adozione di diverse conformazioni.
MODIFICAZIONI POST-TRADUZIONALI: (fosforilazioni, acetilazioni, metilazioni,…).
Proprietà molto importante per tutte le proteine la cui funzione è soggetta a modulazione.
AUMENTO DELLA VELOCITA’ DI INTERAZIONECARATTERISTICHE DEL DOMINIO N-TERMINALE • Il dominio N-terminale è fondamentale per la internalizzazione della proteina (Nunziante, Gilch& Schatzl, 2003), e quindi nel processo di endocitosi, nella regione polibasica formata dai residui aa23– aa28NH2-KKRPKP (Sunyachetal.,2003). • I residui aa23–aa90 sono segnale di bersaglio per i raft ai quali si ancora la proteina tramite l’ancora GPI (Walmsley, Zeng & Hooper, 2003). • La regione polibasica aa23–aa30 sembra cruciale per il corretto ripiegamento della PrPC e potrebbe regolare l’acquisizione della conformazione specifica della proteina patogena (Ostapchenko et al., 2008). • Media un effetto neuroprotettivo (residui aa23-aa50) sia in in vitro che in vivo (Didonna et al.,2012; Flechsig et al.,2003).
IL DOMINIO N-TERMINALE LEGA:
• LEGA LO IONE RAME nella sequenza degli octarepeat ripetuti (residui aa59–aa90), evento
essenziale nel processo di endocitosi (Brown etal.,1997). Delezioni e inserzioni in questa regione
hanno effetto negativo sulla risposta della cellula allo stress ossidativo (Yin et al., 2006; Zeng et
al., 2003) e fanno aumentare la percentuale di resistenza alla digestione da proteasi della proteina
patogena.
• LEGA GLI OLIGOMERI AΒ con alta affinità mediandone l’effetto neurotossico (Parkin, 2007; Chen,
Yadav & Surewicz, 2010; Lauren et al.,2009).
• LEGA LA TUBULINA dei microtubuli del citoscheletro attraverso le regioni nei residui 23-50 e 51-
91 (regione degli octarepeat) e il legame diventa più forte quando il numero di octarepeat aumenta,
dimostrando un ruolo attivo della proteina prionica nella dinamica dei microtubuli all’interno dei
neuroni (Dong, 2008) e provocando un ostacolo al trasporto assonale nel neurone.
• LEGA L’ACETILCOLINESTERASI (AChE), proteina chiave del sistema colinergico nel tessuto nervoso,
è localizzata nei raft lipidici di membrana e nelle terminazioni sinaptiche, come la PrP. L’AChE si lega ai
monomeri PrP (a livello del dominio N-terminale, residui 23-99 e 100-120) e ne induce
l’aggregazione, modificando le proprietà strutturali delle fibrille Prp. Dopo il reclutamento nelle
fibrille PrP, l’AChE perde la sua attività enzimatica e aumenta la citotossicità mediata da PrP. L’AChE
quindi agisce come molecola ausiliaria nel ripiegamento della proteina prionica (Torrent, 2015).IPOTESI SUL MECCANISMO DI CAMBIAMENTO CONFORMAZIONALE DELLA
PROTEINA PRIONICA NEI MAMMIFERI (DOMINIO C-TERMINALE)
• La conservazione attraverso l’evoluzione degli AA nel dominio C-terminale ha fatto pensare che alcuni tratti della
proteina possano essere importanti ai fini della conversione della proteina normale in quella patogena.
• Nel cavallo e nel coniglio (specie resistenti alla infezione da prioni), la strutture della proteina mostra un rigido β2-α2
loop (che per composizione in AA può fornire la possibilità di fornire legami a idrogeno nei foglietti beta) e un
contatto più vicino fra questo loop e la elica α3 (Perez 2010; Wen 2010); quindi la resistenza all’infezione da prioni
sembrerebbe dipendere dalla composizione in AA del loop β2-α2 e dalla sua interazione con l’elica α3 (Legname 2012).
• Infatti, una interruzione del contatto fra il loop β2-α2 e l’ elica α3 nelle proteine mutate (es. Q212P-GSS,V210I-
CJD, E200K-CJD, D178N-CJD-FFI, DOMINIO C-TERMINALE) le rende più flessibili, esponendo maggiormente il
residuo idrofobico e influenzando le proprietà di interazione della proteina con i suoi ligandi; queste modifiche
strutturali della proteina a livello di loop β2-α2 e elica α3 sembrano dimostrare l’esistenza di epitopi critici nella
conversione della proteina da normale a patogena (Meli 2011; Legname 2012).LA MAGGIOR PARTE DELLE MUTAZIONI NEL GENE PRNP (DOMINIO C-TERMINALE)
AUMENTA LA FLESSIBILITA’ DELLA PROTEINA RISULTANTE (MELI 2011)
• Le mutazioni D178N e E208H mostrano un leggero aumento di
flessibilità a livello dei residui 134-156.
• Le mutazioni E196K, ecc. mostrano un aumento di flessibilità fra i
residui 165-175.
• Le mutazioni V180I ecc. mostrano un deciso aumento di
flessibilità in entrambe le regioni comprese fra i residui 165-175
e 185-200.
QUINDI la variazione in flessibilità coinvolge principalmente due
regioni comprese fra i residui 165-175 e 185-200 (dominio C-
terminale) comprendenti il loop β2-α2 e la regione strutturale α2-α3.
L’aumento di flessibilità facilita l’accesso a stati conformazionali
alternativi (Meli et al., 2011), rimodellando i siti della proteina
coinvolti nel riconoscimento dei ligandi e quindi nel riconoscimento fra
molecole.IPOTESI MODELLO DINAMICO (MELI 2011)
Mutazione (sostituzione di un nucleotide con diverso nucleotide)
Sostituzione di un AA con diverso AA Risultato finale:
maggior propensione
della proteina alla
Aumento distanza fra loop β2-α2 e elica α3, per cui ridotta conversione nella
comunicazione fra questi componenti
proteina patogena!!!!
Aumento della flessibilità della proteina
La proteina è costretta a riorganizzare i siti reattivi
coinvolti nel riconoscimento proteina/proteina e
proteina/ligando (es. inattivazione di inibitori o attivazione di
effettori di segnali)
Gli effetti sulla flessibilità della proteina possono essere
trasmessi a tutta la catena proteica, anche a siti distanti
dalla posizione dell’AA mutatoMUTAZIONE P39L: CARATTERISTICHE
DELL’AA PROLINA
• La prolina è un amminoacido non polare, il cui gruppo laterale è
costituito da un anello che non si adatta in una struttura
secondaria ordinata, tendendo ad interromperne la linearità.
• Più proline in sequenza si conformano a loro volta in una tipica
struttura ad elica (POLIPROLINA II (PP II), molto frequente
nelle proteine ad alta flessibilità conformazionale.
• Esistono proteine che incorporano nella loro sequenza molti
residui di prolina. Una di queste è il collagene, proteina
strutturale ubiquitaria nei tessuti animali ed umani. Grazie
alla sua conformazione ad elica, il collagene acquisisce
proprietà fisiche e meccaniche particolari
(elasticità/resistenza alla trazione).
• I domini ricchi di prolina vanno a formare delle "tasche"
molecolari che interagiscono con i ligandi e sono fondamentali
quindi per la trasduzione del segnale intracellulare: infatti la
trasduzione del segnale intracellulare avviene tramite
proteine che possiedono concentrazioni di prolina più
abbondanti rispetto alla sequenza proteica integrale.MUTAZIONE P39L E IPOTESI
SUL RUOLO DEL RESIDUO PROLINA
(DOMINIO N-TERMINALE) (1)
• La regione N-terminale compresa fra i residui 37-53 (poliprolina, PPII) può cambiare nella struttura tramite
idrossilazione in un sito specifico, il residuo 44, in cervello di topi con malattia da prioni (Gill et al., 2000). Ciò
avviene durante o dopo il transito della proteina nel lume del reticolo endoplasmico, grazie al’enzima prolil-4-idrossilasi
(collagene).
• La regione PPII in altre proteine svolge un ruolo regolatorio, permettendo loro di interagire con diversi ligandi a
seconda della conformazione assunta (Siligardi e Drake, 1995; Smith et al., 1997).
• Affinchè avvenga questa idrossilazione (e quindi il cambiamento strutturale) è necessaria una sequenza di consenso
fra i residui 27-29 (Lys-Pro-Gly) e 38-40 (Tyr-Pro-Gly) (Gill et al., 2000).MUTAZIONE P39L
E IPOTESI SUL
RUOLO DEL RESIDUO PROLINA (2)
• E’ possibile che anche per la proteina prionica, come avviene per le altre proteine con
la regione PPII, i cambiamenti della struttura PPII, permettano alla proteina di
variare il riconoscimento di diversi recettori, generando diversi segnali anche con
attività opposte (Tompa et al., 2005, Beland and Roucou, 2012).
• Si è ipotizzato che la mutazione di una prolina in questa regione polibasica (residui
23-50, aumento di flessibilità), critica per la attività neuroprotettiva della PrP (in
risposta allo stress ossidativo), possa permettere una interazione fra proteina e
ligandi più rapida, abolendone le proprietà neuroprotettive e causando effetti
deleteri (Haigh et al., 2009; Turnbaugh et al., 2011).INOLTRE: Gli effetti sulla flessibilità della proteina possono essere trasmessi a tutta la catena proteica, anche a siti distanti dalla posizione dell’AA mutato, causando sia l’assunzione di uno stato semplicemente più ordinato/disordinato, sia cambiamenti di struttura secondaria o terziaria.
QUINDI ESISTE UNA CORRELAZIONE FRA MUTAZIONE DELLA PROTEINA PRIONICA E FENOTIPO SPECIFICO?
MUTAZIONI
Thr183Ala , H187R, Q217R,
Glu196Lys, Pro102Leu, 12 OPRI in
fenotipo FTD-like;
MUTAZIONI
Tyr145X→CAA
Asn171Ser→disordini psichiatrici
Thr183Ala→FAD
OPRI/OPRD→CJD, GSS, AD, HD
Asp
218 Tyr→Asn
(Alzualde et al., 2010)
Jeong BH et al., JKMS 2014NON ESISTE UNA PRECISA
CORRELAZIONE FRA MUTAZIONE DELLA
PROTEINA PRIONICA E FENOTIPO
CLINICO SPECIFICO
Possiamo ipotizzare che il fenotipo FTD-like
clinico e radiologico dipenda dal coinvolgimento
topografico della patologia nelle regioni
frontali prima che in altre regioni.
Questo conferma che la ricerca di mutazioni
nel gene PRNP vada considerata in tali
manifestazioni fenotipiche, in particolare dopo
esclusione delle mutazioni nei geni causativi
FTD.IN LINEA DI MASSIMA ESISTE UNA
PRECISA CORRELAZIONE FRA MUTAZIONE
DELLA PROTEINA PRIONICA E FENOTIPO
NEUROPATOLOGICO SPECIFICO
Infatti, in fenotipi atipici con
mutazioni del gene PRNP per i quali
erano disponibili dati
neuropatologici, questi
dimostravano la presenza di
malattia da prioni (es/ Kumar
2012; Nitrini 2001, Woulfe
2005).MUTAZIONE P39L?
• Il fenotipo clinico FTD-like associato potrebbe dipendere dalla localizzazione della
mutazione in una zona particolare del dominio N-terminale.
• Potrebbe dipendere dalla topografia delle lesioni nelle regioni frontali.
• Non si può comunque escludere che la mutazione P39L sia invece un raro polimorfismo
che predisponga il soggetto a neurodegenerazione.
• Sicuramente sarebbero di grande aiuto la neuropatologia o uno studio su topi
transgenici.GRAZIE PER L’ATTENZIONE!
Puoi anche leggere

















































