Scuola di Specializzazione in Medicina Interna - Direttore: Prof. Paolo Martelletti Canditato: Ottaviani Chiara. Esame finale II AA
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Scuola di Specializzazione in Medicina Interna Direttore: Prof. Paolo Martelletti Canditato: Ottaviani Chiara. Esame finale II AA
CASO CLINICO
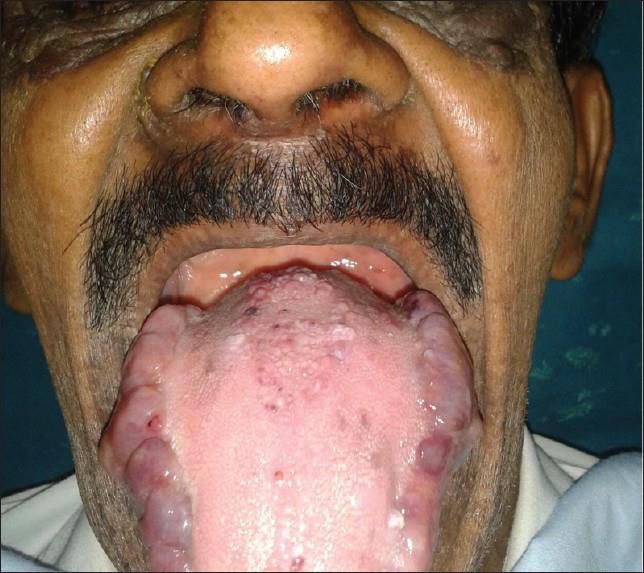
• PC, maschio di 72 aa (medico in pensione, fumatore), giunge in PS per astenia e dispnea per sforzi lievi e
tremori (da Gennaio 2017 ripetuti ricoveri per scompenso). Agli EE grave iponatriemia (120 mEq/l)
• In APR:
• BPCO, ipertensione arteriosa, FA permanente (PMK bicamerale) ipertensione polmonare di
recente insorgenza (post-capillare no tromboembolica), iperomocisteinemia, deficit restrittivo
ventricolo sinistro (già studiato a Pavia), adenoK (resezione anteriore sec Hartmann con
confezionamento di stomia e successiva ricanalizzazione), IRC, vescica neurologica
(autocateterismi)
– ECOCOLOR-DOPPLER CARDIACO: Ipertrofia concentrica, asimmetrica del ventricolo sinistro con
particolare interessamento del SIV. FE 68% … Atrio sinistro ingrandito in assenza di evidenti
formazioni di plus…. Valvola aortica sclerocalcifica … Valvola mitralica insufficienza lieve. Sezioni
destre ipertrofiche con riduzione degli indici di contrattilità ...insufficienza tricuspidalica di se verità
intermedia… Non versamento pericardico. VCI non collassabile .
– ECO-TSA/AAII: ndr
– RX torace: ndr
• Agli EE: nella normaà tsh, ammonio,ca, p, mg, assetto lipidico, C. Difficile negativo, HBV, HCV negativi-
– Alterati: ipergamma policlonale, aumento ggt , bili (diretta), ldh, lipasi, uricemia, IVU , all’EGA alcalosi
metabolica normossiemica normocapnica, cre 2-3, BUN 120-95 , lieve ipoK, all’emocromo PLT
99.000, PCR 1,67, PCT 0,28, trI-HS 46-64, BNP 517-903
• TD in corso: Sintrom, Lasix 500 mg 1/2 cp per 2 ,Arvenum 500 mg 1cp per 2
KCL retard 600 mg 3 cp la mattina, folina 5 mg, Zaroxolyn 1/2 cp, Aldactone 25 mg 2 cp, Allopurinolo 300
mg 1/2 cp
Titolo Presentazione 11/11/17 Pagina 2AMILOIDOSI - definizione
– Gruppo di malattie caratterizzate da un disordine del
folding (ripiegamento tridimensionale) di proteine che,
divenute insolubili, precipitano sotto forma di fibrille
(accumulo spesso in sede extra cellulare).
– Le reazioni sono facilitate da molecole chaperon che si
legano alle proteine → stato di avvolgimento parziale →
cicli di folging-unfolding-folding
– Individuati più di 20 diversi precursori proteici che
possono assumere una conformazione amiloide, motivo
per cui esistono molti diversi tipi di amiloidosi.
– Le fibrille amiloidi insolubili formano depositi
particolarmente stabili a livello di numerosi organi
(alterazioni strutturali e funzionali)
Titolo Presentazione 11/11/17 Pagina 3CLASSIFICAZIONE - 1
ANATOMICA Amiloidosi sistemiche
Amiloidosi localizzate
EZIOLOGICA Amiloidosi primarie
Amiloidosi secondarie
GENETICA Amiloidosi ereditarie
Amiloidosi sporadiche
BIOCHIMICA Tipo di proteina (> 20 tipi diversi)
Titolo Presentazione 11/11/17 Pagina 4CLASSIFICAZIONE - 2
FORMA SISTEMICA: depositi presenti in vari organi (generalmente origine neoplastica,
infiammatoria, genetica o iatrogena). I sintomi e la gravità della malattia dipendono dall'organo
prevalentemente interessato.
Incidenza: 8 casi x milione/anno; M:F 1:1; età: 50-70 anni; prognosi: infausta a 12 anni dalla
diagnosi nell’80% dei pazienti
•Reni: 74%
•Cuore: 60%
•Fegato: 27%
•SNA: 18%
•GI: 8%
•Altro: SNC/SNP, articolazioni, pancreas, milza, cute.
FORMA LOCALIZZATA: confinata ad un particolare organo o tessuto (cuore, reni, tratto
gastrointestinale, sistema nervoso (malattia di Alzheimer), derma ), di solito meno grave.
Titolo Presentazione 11/11/17 Pagina 5A. SISTEMICHE: varianti più comuni
• Amiloidosi primaria (a catena leggera, AL);
• Amiloidosi secondaria (acquisita, AA);
• Amiloidosi ereditaria;
• Amiloidosi sistemica senile (associata
all’invecchiamento).
• è causata da TTR wt
• è più comune nei maschi (già dopo i 60 anni,
ma più frequente dopo gli 80 anni)
• coinvolge preferenzialmente il cuore.
Titolo Presentazione 11/11/17 Pagina 6AMILOIDOSI SISTEMICHE (80-90%)
SEDE FIBRILLA PROTEICA
Discrasie immunocitiche B Rene, intestino, cuore, milza, AL (amiloide a catene leggere)
(plasmacitoma) cute, articolazioni, vie aeree
Amiloidosi reattiva sistemica Rene, fegato, milza, surrene AA (amiloide associata)
(flogosi croniche; LH)
Da emodialisi (insufficienza Osso, cute, legamenti, Aβ2m (β2-microglobulina)
renale) articolazioni
Familiari
Febbre mediterranea (AR) Peritoneo, pleure, membrane AA (amiloide associata)
sinoviali
Polineuropatia (AD) Nervi periferici ATTR (transtiretina met/val 30 o
iso/val122)
Senile Cuore, grandi arterie, ATTR (prevalentemente
polmone, prostata, pancreas normale)
Titolo Presentazione 11/11/17 Pagina 7AMILOIDOSI AL (CATENE LEGGERE)
• Incidenza: 0.8:100.000/anno (è la forma più comune di amiloidosi )
• Fibrille: framento NH2-terminale di mIgG (plasmocitoma a catene
leggere) comprendente la regione variabile + porzione della regione
costante
• Interessa: cuore (75%), reni (65%), fegato (20%), nervi sensitivi
AASS e AAII e SNA (barocettori: 20%). Le cause di morte più
frequenti: insufficienza cardiaca, renale e respiratoria, emorragie,
infezioni.
• Sopravvivenza: 2-4 anni.
• Tp: CHT (melfalan/ciclofosfamide) anti-PCs (comeMM) + DEX. In
alcuni casi autotrapianto SCs previa “bonifica” midollare con melfalan
EV ad alte dosi
Titolo Presentazione 11/11/17 Pagina 8AMILOIDOSI AA (INFIAMMAZIONE CRONICA)
• Amiloidosi secondaria (AA): à acquisita
• condizioni infiammatorie persistenti (TBC, AR, febbre mediterranea
familiare, febbri periodiche ereditarie).
• neoplasie (esempio: Hodgkin’s, CCR).
• CKs pro-infiammatorie (IL-1, IL-6 e TNF) stimolano la produzione epatica di
amiloide serica A (SAA) (concentrazioni sieriche elvate in pazienti con AR,
morbo di Chron … ai flares)
• Depositi tipici: milza, fegato, reni, surreni, linfonodi
• Prognosi : in relazione al trattamento della patologia di base (riduzione dei
valori di SAA); la sopravvivenza è indicativamente di 5-10 anni.
• Tp: sviluppato un farmaco diretto contro i depositi di amiloide, che è in corso di
sperimentazione.
Titolo Presentazione 11/11/17 Pagina 9AMILOIDOSI - PATOGENESI
DISCRASIE IMMUNITARIE REATTIVE
STIMOLO Cause ignote Infiammazione cronica
Proliferazione LB Attivazione macrofagi
Plasmacellule IL 1-6
PRECURSORE
SOLUBILE Catene leggere Ig Proteina SAA
Proteolisi parziale
FIBRILLE
INSOLUBILI AL AAAMILOIDOSI LOCALIZZATE (10-20%)
SEDE FIBRILLA PROTEICA
Endocrine
Carcinoma tiroideo Tiroide ACal (calcitonina)
Insulinoma Pancreas AIAPP (amilina)
Diabete Tipo II Pancreas AIAPP (amilina)
Amiloidosi isolata atriale Atri AANF (fattore natriuretico atriale)
Cerebrali
M. Di Alzheimer Neocorteccia, ippocampo, Aβ (APP)
amigdala
Encefalopatia spongiforme Neuroni cerebellari APrP
HCHWA I (AD) Arterie/arteriole cerebrali e Cistatina C (glu/leu68)
leptomeningee
HCHWA O (AD) Arterie/arteriole cerebrali e Aβ (APP)
leptomeningee
Titolo Presentazione 11/11/17 Pagina 11Possibili effetti dei depositi fibrillari
Organo o sistema Possibili conseguenze
interessato
Cervello Malattia di Alzheimer
Apparato Macroglossia, disfagia, diarrea, stipsi, ostruzione intestinale,
digerente malassorbimento
Cuore Aritmie/ insufficienza cardiaca
Reni Edemi , proteinuria, IRC
Fegato Epatomegalia
Polmoni Dispnea
Sistema nervoso Sindrome del tunnel carpale, parestesie, disestesie
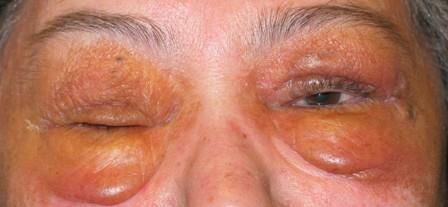
Pelle Papule, porpora ed ecchimosi
Tiroide Gozzo
Titolo Presentazione 11/11/17 Pagina 12AMILOIDOSI EREDITARIE
• Gruppo eterogeneo di patologie (ereditarietà AD a penetranza variabile)
• Una o più mutazioni in una proteina specifica
• Manifestazioni sistemiche/tessuti preferenziali
• Sistema nervosoà caratteristica neuropatia simmetrica sensitivo-motoria
AAII.
• Cuore, vasi, reni.
• ATTR (cuore/nervi periferici) e AApoA1 (fegato,rene, cuore): le due forme più
comuni in Italia
• Diagnosi: DNA
• La prognosi varia con il tipo di mutazione genica e il grado di avanzamento al
momento della diagnosi.
•Tp: disponibili o in studio nuovi farmaci che bloccano la formazione dei depositi di
amiloide o che promuovono la dissoluzione dei depositi già formati.
Titolo Presentazione 11/11/17 Pagina 13SIGLA CLASSIFICAZIONE:
PRECURSORE AMILOIDOSI FAMILIARE - 1
CLINICA
PROTEICO
FORME SISTEMICHE
ATTR Transtiretina Amiloidosi familiare e senile (polineuropatia)
APO-A1,2 Apolipoproteina-A 1,2 Amiloidosi familiare renale (sd. nefrosica)
AFib Fibrinogeno Amiloidosi familiare renale (Ostertag)
AGel Gelsolina Amiloidosi familiare
ALys Lisozima Amiloidosi familiare
ACyst Cistatina C Amiloidosi familiare
FORME LOCALIZZATE
ABri ABriPP Demenza, familiare
ADan ADanPP Demenza, familiare
Aker Cheratoepitelina Cornea, familiare
ALac Lattoferrina Cornea, familiare
Titolo Presentazione 11/11/17 Pagina 14CLASSIFICAZIONE: AMILOIDOSI FAMILIARE - 2
A TTR (transtiretina)
AMILOIDOSI SECONDARIA
AL – catene leggere
AA – Flogosi croniche A ApoAI
b2-microglobulina
A Gel (gelsolina)
…
ALTRE FORME
NON TTR
• A Lys (lisozima)
• A Cys (cistatina C)
• A Fib (fibrinogeno catena Aa)
• A Apo AII (apolipoproteina AII)
• … (>20 Pr amiloidogenetiche nell’uomo)
Titolo Presentazione 11/11/17 Pagina 15Transtiretina (TTR)
• Proteina plasmatica a struttura tetramerica ® subunità
identiche di 127 AA
• Sintetizzata dal fegato; presente anche nel cervello (plesso
corioideo) e nell’occhio (epitelio pigmentoso della retina)
• Trasporta: RBP e ¼ della tiroxina sierica
• E’ codificata da un gene di 4 esoni sul CR 18:
• esone 1 ® 20 AA solo 3 nella forma matura
• esone 2 ® 4-47 AA
• esone 3 ® 48-92 AA
• esone 4 ® 93-127 AA
Titolo Presentazione 11/11/17 Pagina 16Transtiretina (TTR)
• > 80 mutazioni amiloidosiche identificate (carattere AD)
• SNPs del gene TTR (Cr18):
no mutazioni per esone 1;
37 per esone 2;
45 per esone 3;
16 per esone 4
• Neuropatia amiloidosica da mutazioni di TTR
Titolo Presentazione 11/11/17 Pagina 17Transtiretina
Familial Amyloid Polineuropathy (FAP 1)
• Precoce: SNP (sensoriale, motoria con progressione
centripeta prevalentemente AAII) e SNA
• Polineuropatia S-M progressiva (parestesie, dismot.
ingreavescenti), disfunzioni GI e urinarie (alvo alterno,
CIP, ritenzione), impotenza, ipotensione ortostatica
• Raramente depositi di A . meningei e cerebrovascolari
Titolo Presentazione 11/11/17 Pagina 18Apolipoproteina A-1
• Apo A1
• 243 AA
• 12 mutazioni (generalmente sostituzioni di singoli
nucleotidi) sul Cr 11 ® frammenti di Apo A1 ®
degradazione incompleta ® deposizione di residui 83-
93 sotto forma di fibrille
• Ereditarietà AD a penetranza variabile (insorgenza, severità)
• Mutazioni C-terminali ® modulazione del metabolismo
della proteina: deposizioni renali (interstiziali), epatiche e
cardiache
• Mutazioni N –terminali ® deposizioni cutanee e laringee
• APO-A1 mutazione Glyc26Arg àneuropatia periferica
Titolo Presentazione 11/11/17 Pagina 19Gelsolin (A Gel) • Sintetizzata nel muscolo scheletrico e nei macrofagi; gene sul Cr 9 ® clivaggio di actina (essenziale x metabolismo / riorganizzazione citoscheletrica) • 2 mutazioni associate a deposizione di A composta di un segmento interno di gelsolina costituito da 71 AA comprendenti il residuo mutato (Asp187Asn, Asp187Tyr) • Distrofia corneale, neuropatia NNCC, lassità cutanea del volto – Asp187Asn Titolo Presentazione 11/11/17 Pagina 20
Lisozima (A Lys) • Proteina di 14.500 Dalton con proprietà batteriolitiche (indice di flogosi, anche in linfomi / leucemie) • 4 mutazioni associate a deposizione di A composta di vari segmenti, ma anche della proteina intatta • Deposizione di A nei glomeruli ® IRC tardiva (insorgenza 3/4° decade di vita) • Trapianto di rene frequentemente efficace
AMILOIDOSI – sospetto quando…. • Qualsiasi paziente con malattia sistemica multiorgano senza causa apparente (anamnesi!) • CLINICA/EO: edemi, dispnea, >creat-BUN, proteinuria, > NT- proBNP, BNP e troponina, epato/splenomegalia,
Titolo Presentazione 11/11/17 Pagina 23
AMILOIDOSI – diagnosi • DD: miocardiopatie, neuropatie (TTR), IRC (Apo A1, Apo A2) …………. • Biopsia colorata con Rosso Congo (da organo affetto oppure grasso periombelicale o mucosa rettale ® sensibilità 70-80% in TTR) • Test genetici x le forme ereditarie Titolo Presentazione 11/11/17 Pagina 24
Puoi anche leggere

















































