INSEGNAMENTI DALLO STUDIO DELLE MALATTIE RARE - Dott. Andrea Bartuli UOC Malattie Rare e Genetica Medica Dipartimento Pediatrico Universitario ...
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù

INSEGNAMENTI DALLO
STUDIO DELLE
MALATTIE RARE
Dott. Andrea Bartuli
UOC Malattie Rare e Genetica Medica
Dipartimento Pediatrico Universitario Ospedaliero
Ospedale Bambino Gesù, Roma
MALATTIE COMUNI DELL’INFANZIA
1950
PATOLOGIE DELL’ACCRESCIMENTO
PATOLOGIE NUTRIZIONALI
BENESSERE
ANTIBIOTICI PATOLOGIE ENDOCRINE
VACCINAZIONI MALATTIE INFETTIVE
STRUMENTI DIAGNOSTICI
2016
MALATTIE RARE DELL’INFANZIA
2016
PAZIENTI METABOLICI
ONCOEMATOLOGICI
SCREENING
CHIRURGIA PAZIENTI MALFORMATI
GRANDI PREMATURI
NUTRIZIONE
RESPIRAZIONE
TRAPIANTOLOGIA 1980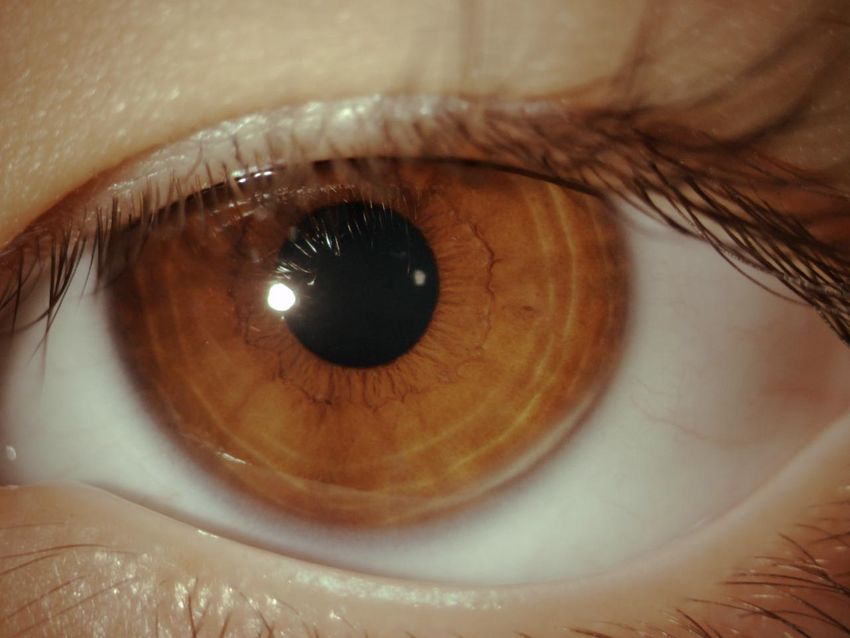
• SOPRAVVIVENZA A CONDIZIONI LETALI IN EPOCA NEONATALE + • ASSISTENZA PAZIENTI CON GRAVI PATOLOGIE APPARATO DIGERENTE + • ASSISTENZA PAZIENTI CON GRAVI PATOLOGIE APPARATO RESPIRATORIO + • CRONICIZZAZIONE DI PATOLOGIE ACUTE + • SENSIBILITA’ DIAGNOSTICA PER MALATTIE RARE = 0-16 a PREVALENZA DI MALATTIE DISABILITANTI 1:200 PERSONE AFFETTE DA MALATTIE RARE # 1 ogni 20
Una Malattia è Rara se ha frequenza non > 1:2000 Le MR sono più di 8000 ITALIA 2 milioni di pazienti affetti (1700000 < 16 aa) EUROPA 20 milioni di pazienti affetti Malattie complesse, evolutive, Multiorgano

80 % eziologia genetica del 45% delle malattie rare conosciamo i geni responsabili e i meccanismi eziopatogenetici nel 50 % esordio prima dei due anni nel 70 % esordio in età pediatrica
Causa di morte: < 12 mesi 33 %
1 - 5 anni 10 %
5 -15 anni 12 %
70% dei pazienti non riceve una diagnosi prima di due anni
40% dei pazienti resta senza diagnosi
65 % esiti invalidanti
44% disabilità motoria
37% mutilazioni sfiguranti
per il 30% dei pazienti è disponibile
un trattamento in grado di
modificare significativamente la
storia clinica naturale
ogni paziente ha bisogno nella sua
vita di 5 specialisti diversi
Non diagnosticate
45-35% 1:2.000.000
Ultra -Rare
55-65% Marshall Smith 1:1.000.000
Rubinstein Taybi 1:100.000
Cornelia de Lange 1:50.000
Riconoscibilità
Smith Magenis 1:25.000
Angelman 1:20.000
CHARGE 1:10.000
Down 1:1.000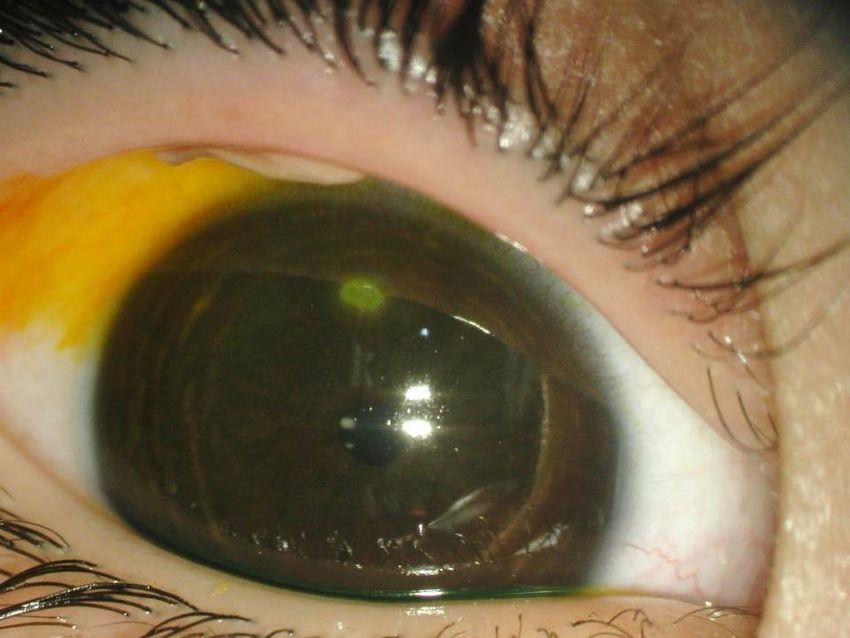
TUMORI SOLIDI E LIQUIDI
MALATTIE ENDOCRINE
MALATTIE METABOLICHE
NON DIAGNOSTICATE
MALATTIE RARE
MALATTIE INFETTIVE
SINDROMI GENETICHE
MALATTIE NEUROLOGICHE
MALATTIE PSICHIATRICHE
PATOLOGIE AMBIENTALIPECULIARITA’ SOCIALE DELLE MALATTIE RARE
1 BAMBINO AFFETTO = 4 GENERAZIONI COINVOLTE
NONNI
GENITORI
FRATELLI
FIGLII GENITORI E IL BAMBINO CON MR CONFLITTO TRA: RUOLO GENITORIALE (responsabilità emotiva) E RUOLO DI CASE MANAGER (responsabilità clinica) ABBANDONO DEL LAVORO ELEVATO TASSO DI SEPARAZIONI
IL PEDIATRA E IL BAMBINO CON MR - SOTTOSTIMA DEI SINTOMI/RIFIUTO - ASSENZA DI LINEE GUIDA DI DIAGNOSI E FOLLOW-UP - ASSENZA DI RETE SUL TERRITORIO
L’OSPEDALE E IL BAMBINO CON MR - PROTOCOLLI DI FOLLOW-UP SOLO PER LE MR FREQUENTI - RICOVERI INAPPROPRIATI - COMPLESSITA’ DEI RICOVERI - ASSENZA DI COLLEGAMENTO CON IL TERRITORIO
PECULIARITA’ ASSISTENZIALE DELLE MALATTIE RARE
MALATTIA COMUNE MALATTIA RARA
monosintomatiche multiorgano
1 malattia=1 medico/ospedale 1 malattia=molti medici/ospedali
diagnosi (frequente) diagnosi (70%)
cura cura nel 30%
guarigione stato di malattia cronica
o evoluzione di disabilità
andrea.bartuli@opbg.net- 1997 Stati Uniti, tutti gli organismi impegnati nelle MR vengono raccolti in un'unica organizzazione: il NORD (National Organization for Rare Disorders) - 1999-2003 Programma d'Azione Comunitaria sulle MR nel quadro dell’Azione della Sanità Pubblica, Parlamento e Consiglio Europeo - 2000 Regolamento concernente i medicinali orfani Istituzione della procedura comunitaria per la qualifica di medicinale orfano Istituzione EMA (European Medicines Agency) e COMP (Committee for Orphan Medicinal Products)
ISTITUZIONE DELLA RETE NAZIONALE DELLE MALATTIE RARE (D.M. 279/2001) DEFINISCE la Rete Nazionale per la prevenzione, la sorveglianza, la diagnosi, la terapia delle MR ISTITUISCE il Registro Nazionale delle MR presso l'ISS REGOLAMENTA l’esenzione dalla partecipazione al costo delle malattie elencate
- sviluppo di azioni di prevenzione, sorveglianza, migliorare diagnosi e terapia, promuovere l’informazione e la formazione, ridurre l’onere che grava sui malati e sulle famiglie - creazione di una rete costituita da presidi accreditati, appositamente individuati dalle Regioni per erogare prestazioni diagnostiche e terapeutiche
- Registro Nazionale Malattie Rare per avere dati sulla prevalenza e l’incidenza - Definizione di 47 gruppi di malattie comprendenti 284 patologie (congenite e acquisite) ai fini dell’esenzione dalla partecipazione al costo delle prestazioni sanitarie correlate - Promozione di protocolli diagnostici e terapeutici comuni, sviluppo attività di ricerca
ACCORDO STATO-REGIONI 2007 Il Governo, le Regioni e le Province autonome di Trento e di Bolzano convengono: - “che le Regioni si impegnano ad attivare i registri regionali sulle malattie rare entro il 31.03.2008 e a garantire il collegamento con il registro nazionale presso l’ISS” - “che il Registro nazionale produca le evidenze epidemiologiche a supporto della definizione e dell’aggiornamento dei Livelli Essenziali di Assistenza, nonché delle politiche e della programmazione nazionale”
DIAGNOSI
DIAGNOSI FENOTIPICA
DIAGNOSI BIOCHIMICA
DIAGNOSI GENETICA
PATOLOGIE CROMOSOMICHE PATOLOGIE MENDELIANE
Sindrome di Down Fenilchetonuria
Sindrome di Turner Intolleranza ereditaria al fruttosio
Sindrome di Klinefelter Ipercolesterolemia familiare
Sindrome di Edwards Emofilia
GENOMA AMBIENTEIl sequenziamento dell’Esoma
• Analizza la parte del genoma meglio compreso
(esoni – gene-- proteina)
• Esoni comprendono circa 1% del genoma
• ~ 85% di tutte le malattie conosciute causate da
mutazioni si trovano su esoni
• Costo di sequenziamento dell'esoma: 1/6 del costo del
sequenziamento dell'intero genomaPatologie per cui applicare l’analisi Esomica • Anomalie congenite multiple • Disabilità intellettiva • Regressione
Analisi di sequenziamento e workflow bioinformatico
~ 60 000 ÷ 70 000 DNA VARIANTS
Discrete filtering
NONSYNONYMOUS, NONSENSE,
FRAMESHIFT, SPLICE SITE
Discrete filtering
ABSENT IN UNAFFECTED
POPULATION CONTROLS
in-house DB
Discrete filtering
FUNCTIONAL ANNOTATION
Discrete filtering
SEGREGATION ANALYSIS
(pedigree information, models of inheritance)
~1÷5
Prioritization Prioritization
A PRIORI
Making Sense of Data KNOWLEDGESequenziamento dell’Esoma nella Pratica Clinica - Pazienti non selezionati, senza diagnosi clinica, negativi ad analisi citogenetiche e molecolari - Diagnosi ottenuta nel 25- 28% dei casi esaminati 40% NON DIAGNOSTICATI + 10 % DIAGNOSTICATI
UDP15001
TELETHON UNDIAGNOSED
DISEASES PROGRAM
three clinical sites and a coordination center
• Coordinator
Vincenzo Nigro, Sandro Banfi
• Partner 1
Bruno Dallapiccola,
Marco Tartaglia
• Partner 2
Angelo Selicorni,
Andrea Biondi
• Partner 3
Nicola Brunetti Pierri,
Giancarlo Parenti
This program is an intramural effort of Telethon centered on the TIGEM (Pozzuoli)
where NGS activities will be converged and will rely on a core network of three
centers with great expertise in clinical genetics and pediatricsè diretto…
a 350-400 famiglie con bambini senza diagnosi genetica né clinica
esempi: malformazioni a carico di diversi organi ed apparati, difetti congeniti del
metabolismo, malattie neurologiche o neuromuscolari, disabilità intellettiva
sindromica non nota, gravi difetti della vista o dell’udito, ecc
non è diretto…
• a chi non ha una malattia genetica
• a chi ha una malattia genetica nota, ma ha non ha ottenuto ancora una diagnosi
molecolare
• a chi ha una condizione parzialmente genetica (schizofrenia, autismo, ritardo di
crescita, sclerosi multipla, patologia neoplastica, epilessia, etc)
• a chi ha un’anomalia cromosomica
• a malattie dell’età avanzata o disabilità intellettiva isolataLa campagna sociale Vite Coraggiose, promossa dalla Fondazione Bambino Gesù
- e lanciata in occasione del Giubileo Straordinario della Misericordia indetto da Papa
Francesco - è un progetto teso a raccogliere fondi a sostegno della ricerca e cura delle
malattie rare e ultra rare, di cui l’Ospedale Bambino Gesù è centro di riferimento.
Circa il 5% di tutti malati rari registrati in Italia viene diagnosticato al Bambino Gesù.
Parliamo di oltre 10.000 bambini ogni anno, la più ampia casistica nazionale in ambito
pediatrico.
Queste cifre, di per sé significative, non registrano tuttavia la parte sommersa dei malati
ultra-rari, circa la metà dei casi, rappresentata da quei bambini la cui condizione resta
senza ipotesi diagnostica.
Vite Coraggiose è una campagna nazionale di sensibilizzazione tematica che parla del
coraggio, della forza e della determinazione che accomuna ricercatori, medici, famiglie e
bambini nella medesima battaglia per la qualità della vita. Se non per la vita stessa.
Tanto più quando si parla di malattie di cui a volte non si conosce nemmeno il nome.
Obiettivo: costruire per migliaia di bambini ultra-rari, orfani di diagnosi, l’inizio di un
percorso in grado di accompagnarli dalla definizione della loro condizione clinica alla
cura della loro patologia.
Fondazione Bambino GesùWorkflow del progetto OPBG:
reclutamento della casistica
Selezione clinica della casistica
GENETICA MEDICA/MALATTIE RARE
raccolta anamnestica e implementazione delle informazioni cliniche mediante la
richiesta di indagini strumentali, morfologiche e cognitivo-comportamentali
Discussione casi clinici non diagnosticati
REVISIONE INTERNA
Assicurare che il paziente soddisfa i criteri per essere inserito nel progetto Exome,
Diagnosi differenziale, analisi di geni malattia etc
INVIO CAMPIONE PER ANALISI INVIO CAMPIONE PER
GENI CANDIDATI ANALISI ESOMICANuovo sottotipo di sindrome con Blefarofimosi-Ptosi e Disabilità Intellettiva
Am J Med Genet, 2011
Blefarofimosi, Ptosi, telecanto, rime palpebrali vs l’alto
Microcefalia (pre-natale), Ritardo mentale di tipo
grave
Capelli radi e sottili
Ipotonia/ iperlassità
Am J Human Genet 2012; 91:998-1010 Difficoltà nell’alimentazione/ scarso accrescimento
Anomalie congenite multiple (cardiaco, GI, renale)
Ipoplasia del corpo calloso
Bassi livelli di colesteroloSindrome di Zimmermann- Laband
Malattia rara caratterizzata da deficit cognitivo, fibromatosi gengivale, assenza/ipoplasia delle unghie e delle
falangi terminali delle mani e dei piedi, ipertricosi, e in alcuni pazienti, da epilessia
Nat Genet. 2015 Jun;47(6):661-7Sindrome di Aymè- Gripp
Malattia rara caratterizzata da cataratta congenita, sordità neurosensoriale, ritardo mentale, bassa statura,
epilessia e da una facies caratteristica
Gene MAF
Am J Hum Genet. 2015 May 7;96(5):816-25Sindrome di Noonan
In corso analisi
esomica su
pazienti
selezionati con
diagnosi di
RASopatia,
negativi ai geni
notiDalla teoria….. …alla pratica
NON DIAGNOSTICATE 800mila
Claudia marzo 2006 Primogenita di genitori non consanguinei, anamnesi familiare non significativa, gravidanza decorsa senza problemi, Parto eutocico alla 40° settimana, PN 3,550 kg, L 51 cm, cc 34 cm
Sviluppo neuromotorio con lieve ritardo delle tappe evolutive, seduta a 9 mesi, cammina a 20 mesi 1 anno e 9 mesi RMN encefalo NORMALE (“Non evidenti aree di alterata intensità di segnale nei tessuti cerebrali in sede sovra e sottotentoriale. Non si rilevano aspetti patologici in regione ippocampale paraippocampale. Non evidenza di alterazioni della migrazione né difetti della solcazione. Normale il grado di mielinizzazione in rapporto all’età. Normale visualizzazione degli spazi sub aracnoidei della base e del mantello. Sistema ventricolare in asse, di regolari morfologia e dimensioni. Lieve dilatazione del IV ventricolo. Dilatazione della porzione intracranica della cisterna magna per megacisterna magna. Normale rappresentazione del corpo calloso. Normoposizione delle tonsille cerebellari. Non aspetti patologici in sede tronco-encefalica. La sequenza DWI non dimostra alterazioni della diffusione a carico dei tessuti cerebrali”)
2006-2015 Valutazioni cliniche: 6 Genetiche da 4 diversi Genetisti 4 Oculistiche da 3 diversi Oculisti 4 Neuropsichiatriche da 2 Nps 21 Neurologiche da 8 diversi Neurologi 6 Metaboliche da 3 diversi Pediatri Ricoveri: 2 Ospedali (regionali) 2 Cliniche Universitarie (regionale, extraregionale)
Esami strumentali: 2 RMN encefalo (1 spettroscopica) 4 ecografie addome 1 radiografia mano e polso 3 Studi Neurofisiologici (BAEP, PEV, PEU) Esami biochimici: 5 screening metabolici (3 Ospedali) 1 studio assetto vitaminico 47 prelievi per studio assetto immunitario, endocrino, parametri biochimici, indici infiammatori, infettivi, emocolture, etc 7 esami genetici (Cariotipo standard, CGH-array, mutazione celiachia, gene SHANK3, MTHFR, pannello epilessie, pannello cervelletto)
Claudia viene proposta nel gennaio 2015, ha 9 anni e 10 mesi Studio esomico: presenza di mutazione sul cromosoma 22 significativa per gene dell’Adenilsuccinato liasi (ADSL) OMIM #103050, *608222 Studio genetico: Mutazione missense (a) = c.926 G>A esone 9, p.R309H (trasmessa dalla madre) Mutazione di splice ( b) = IVS11+5 G>C (trasmessa dal padre) Studio enzimatico: Attività ADSL Claudia Roma = 80.95 UI/l eritrociti Attività ADSL controlli = 320 – 550 UI/l eritrociti Esami Metabolici per ADSL: AICAr 1.72 assente SAICAr 57.59 assente SUCCINILADENINA 4.11 assente
COSTO RITARDO DIAGNOSTICO IN 8 anni Economico - costo indagini diagnostiche/ricoveri 72mila euro - Costo studio esomico 800 euro Emotivo Rischio riproduttivo - 1 gravidanza
What is the Undiagnosed Diseases Network? The Undiagnosed Diseases Network (UDN) is a research study that is funded by the National Institutes of Health Common Fund . Its purpose is to bring together clinical and research experts from across the United States to solve the most challenging medical mysteries using advanced technologies. Through this study, we hope to both help individual patients and families and contribute to the understanding of how the human body works.
“At the age of 2 years old, Asia began our medical journey with 2 tonic-clonic seizures. Over
the next several years, her list of medical symptoms grew and grew. We were often told by
her doctors that she would “grow out” of many of her symptoms but we felt it was more
than that, so we kept testing her for a “diagnosis”. Asia had every test available to us
including lumbar punctures, biopsies, MRI’s, & blood work. We also saw several different
types of doctors including specialists and natural healing. No diagnosis could be found. Five
years later at the age of 7, her neurologist put in a request for a genetic test called Whole
Exome Sequencing study. He was very reluctant that this test would be approved by our
insurance company but it was and we went forward. After a simple blood draw, we had our
answer just a few short days later. Asia was diagnosed with a rare genetic disorder.
I was confused and angry when I received this result. I had put
her through so much pain & discomfort looking for this answer
and it came with a single test. Why did I do that to her, why was
this not an option earlier on, why won’t insurance companies pay
for genetic testing, and why is this not available to everyone
needing it when it could save lives and so much heartache?
Now we had the diagnosis, what do we do?In 2014, the NIH Undiagnosed Diseases Program will expand to include a network of major academic medical centers across the U.S. The Genzyme/NORD NIH Undiagnosed Diseases Fund will support testing for patients applying to all clinical sites and, in particular, the new satellite centers. Patients applying to the NIH program who need assistance to pay for the required testing will be referred to NORD by the NIH centers. Nel 2015 stabilisce che “ogni paziente non diagnosticato dopo 6 mesi in carico a struttura di III livello, dovrebbe essere sottoposto a studio esomico”.
ULTRARARE 1:1milione
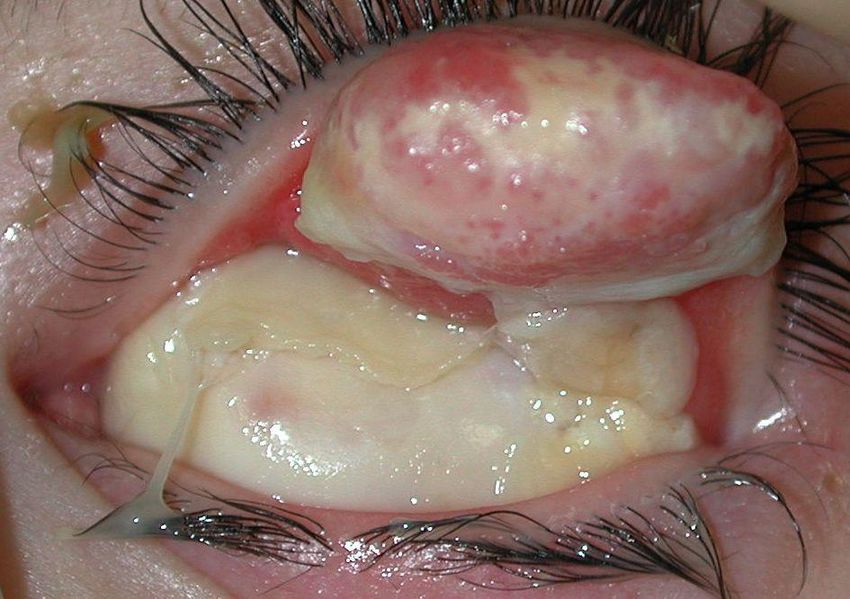
Giulia 5 anni Genitori non consanguinei, anamnesi familiare non significativa Dall’età di 2 anni congiuntiviti ricorrenti con comparsa di lesioni palpebrali (calazio?) Trattamento: antibiotici e steroidi topici e 2 escissioni chirurgiche con recidiva dopo poche settimane e peggioramento delle lesioni. andrea.bartuli@opbg.net
Occhio destro
Pseudomembrana: colorito bianco-giallo, consistenza morbida
localizzata congiuntiva tarsale superiore.
Piccola area di disepitelizzazione corneale con opacità stromale
paracentrale (fluoresceina+)
andrea.bartuli@opbg.netOcchio sinistro Pseudomembrana colorito rosso, consistenza lignea, 9 mm altezza, adesa al bordo palpebrale superiore e alla congiuntiva tarsale, ectropion palpebrale. Membrana di colorito giallo-bianco, congiuntiva bulbare, fornici e cornea. Tisi bulbare. RMN: Distacco retinico (secondario a endoftamite)
Sospetto diagnostico:
Congiuntivite lignea/Deficit Plasminogeno
• Trasmissione autosomica recessiva
• Malattia ULTRARARA SISTEMICA
(>1/1milione in omozigosi o eterozigosi composta)
• Ridotta sintesi di plasminogeno con ridotta FIBRINOLISI
e formazione di PSEUDO-MEMBRANE di consistenza
LIGNEA durante il processo di RIPARAZIONE delle lesioni
mucosali
andrea.bartuli@opbg.netDeficit di Plasminogeno di tipo 1 SISTEMICA andrea.bartuli@opbg.net
Diagnosi biochimica
• giulia plasminogeno plasmatico < 29% (vn > 70%)
• madre 72%
• padre 115%
Diagnosi Genetica (Lipsia University)
singola mutazione (K19E) gene PLG ereditato dal padre
Trattamento sperimentale
Plasminogeno umano topico (Kedrion, Lucca) veicolato in
acido ialuronico (Watts P et al. Am J Ophthalmol. 2002 Apr;133(4):451-5)
2 gocce ogni 2 ore in entrambi gli occhi
andrea.bartuli@opbg.netOcchio destro
Prima del trattamento Dopo 7 giorni
andrea.bartuli@opbg.netOcchio sinistro
Prima del trattamento Dopo 7 giorni
andrea.bartuli@opbg.netTrattamento
chirurgico
Applicazione
protesi
andrea.bartuli@opbg.netFollow-up a 12 mesi
Assenza di membrane e di recidive anche dopo
trauma chirurgico e di protesizzazione
andrea.bartuli@opbg.netFollow-up a 6 anni Assenza di comparsa di nuove lesioni sia nell’occhio protesizzato che in quello sano Schema posologico e costi 1 gtt ogni 2 ore Plasminogeno Kedrion uso compassionevole Acido ialuronico (Biolon) 25 euro a fiala, 3 fl/die, 55.000 euro/anno (consegna mensile, ricovero ordinario 24.000, euro anno) 1 gtt ogni 4 ore Plasminogeno Kedrion uso compassionevole Acido ialuronico (Biolon) 25 euro a fiala, 3 fl/die, 27.350 euro/anno (consegna mensile, ricovero diurno, 3,600 anno) 1 gtt ogni 4 ore Plasminogeno Kedrion uso compassionevole Acido ialuronico (Dropstar) 0,47 a fiala, 2 fl/die, 353 euro/anno (consegna mensile, ambulatorio, 240 euro anno) Conforti FM, Di Felice G, Bernaschi P, Bartuli A, Bianco G, Simonetti A, Buzzonetti L, Valente P, Corsetti T. Novel plasminogen and hyaluronate sodium eye drop formulation for a patient with ligneous conjunctivitis. Am J Health Syst Pharm. 2016 Apr 15;73(8):556-61.
- PDTA solo per le MR più frequenti - Evidence Based Medicine Hope Based Medicine - Ricoveri inappropriati, Ambulatori non remunerati - Assenza di collegamento ospedale/territorio - Copertura assistenziale dei bisogni delle famiglie con disabilità del 30% (55% Nord Italia -8% Sud Italia) - Disparità intraregionale
CTs in EU
CTs in ADULT population CTs in PAEDIATRIC population
5000
4500
4000
3500
3000
2500
2000
1500
1000
500
0
2005 2006 2007 2008 2009 2010 2011
(EudraCT data)Farmaci in pediatria
ADULTI BAMBINI
▪ Farmaci approfonditamente ▪ 20% popolazione EU (100 mil): < 16
aa → prematuri, neonati a termine,
studiati bambini, adolescenti
▪ 50-90% medicinali utilizzati in
pediatria NON è appositamente
studiato
✓ Sicurezza Rischi:
✓ Efficacia ✓ ADRs (sovradosaggio)
✓ Alta qualità ✓ inefficacia (sottodosaggio)
✓ formulazioni inadeguate
✓ Scoperta di farmaci innovativi ✓ ritardi nell’accesso a farmaci
innovativiUso inappropriato dei medicinali approvati
• 3/4 medicinali in commercio oggi NON supportati da
indicazioni approvate da EMA/FDA per l’uso nei neonati,
lattanti, bambini e adolescenti
• Aziende Farmaceutiche non supportano la ricerca necessaria
per lo sviluppo dei farmaci pediatrici → scarso mercato
• Negli SPC spesso riportati “disclaimers” → sicurezza e
l’efficacia non sono state specificatamente studiate
“Uso Off-Label” (≠ uso scorretto!)
Pazienti pediatrici in “sperimentazione” per qualsiasi uso
terapeutico non registrato
Assenza di dati basati su evidenze che supportino come
somministrare il farmaco in modo sicuro ed efficaceLO SVILUPPO DI UN FARMACO
STUDI PRE-CLINICI STUDI CLINICI FASE REGISTRATIVA
FASE I preliminari Richiesta di
Scoperta e selezione
sicurezza commercializzazione
delle molecole
(soggetti sani, ~20-80)
FASE II Generazione di Ipotesi
Studi su animali Valutazione delle autorità
Sicurezza, PK, Efficacia
sanitarie (EMEA)
(pazienti, ~100-200)
FASE III Confermativi
Richiesta autorizzazione (pazienti, ~1000-3000)
alla sperimentazioneORGANIZZAZIONE SANITARIA TRADIZIONALE
neurologia endocrinologia genetica metabolismo broncoORGANIZZAZIONE MULTIDISCIPLINARE
neurologia endocrinologia M.RARE metabolismo broncoAMBULATORIO modello tradizionale
AMBULATORIO multidisciplinare
RARE >1:2mila
Filippo
3 anni
- Anamnesi familiare positiva ipercolesterolemia e MCVp
- Ai 2 aa, presso altro centro, C-LDL di 607 mg/dl. Studio genetico
dimostrativo di eterozigosi FH.
- Trattato con inibitori dell’assorbimento del colesterolo senza
miglioramento.
- A 3 aa valutazione OPBG.
C-tot 594 mg/dl (v.n. 120-200)
C-HDL 39 mg/dl
C-LDL 533 mg/dl (v.n. 50-150)
Indagini strumentali (ecocardiogramma, ecografia tendinea,
ecografia epatica, ecodoppler vasi collo, coronarografia) negative
andrea.bartuli@opbg.netIpercolesterolemia Familiare Ridotto catabolismo o aumentata sintesi di LDL-C con aumento della sua concentrazione ematica e deposito progressivo nei vasi e nei tessuti Trasmissione autosomica dominante ETEROZIGOSI 1:380 (malattia comune) OMOZIGOSI 1:780000 (malattia rara) Maiorana A, Nobili V, Calandra S, Francalanci P, Bernabei S, El Hachem M, Monti L, Gennari F, Torre G, de Ville de Goyet J, Bartuli A. Preemptive liver transplantation in a child with familial hypercholesterolemia. Pediatr Transplant. 2011 Mar;15(2):E25-9.
Ipercolesterolemia Familiare-Eterozigosi Sintomatologia clinica: - xantomi a comparsa dopo l’età pediatrica, malattia CV dopo 30 anni (♂ 42-46 aa, ♀ 51-52aa) Esami biochimici, LDL-C: - 30 aa ≥250 mg/dl
Ipercolesterolemia Familiare-Omozigosi
Sintomatologia clinica:
- xantomi a partire dai 18 mesi (osservazione personale),
arcus cornealis
- 20 aa età media di comparsa di MCV (5 anni,
osservazione personale)
Esami biochimici
LDL-C >600 mg/dl
Macchiaiolo M, Gagliardi MG, Toscano A, Guccione P, Bartuli A.
Homozygous familial hypercholesterolaemia.
Lancet. 2012 Apr 7;379(9823):1330.Ipercolesterolemia Familiare Omozigote (oIF)
Macchiaiolo M, Buonuomo PS, Valente P, Rana I, Lepri FR, Gonfiantini MV, Bartuli A. Corneal arcus as first sign of familial hypercholesterolemia. JPediatr 2014 Mar;164(3):670
Studi genetici (Prof. S.Calandra, Università di Modena):
Fenotipo omozigote (eterocomposto) dovuto alla presenza di due
mutazioni
- la sostituzione della Valina in posizione 502 con una
Metionina su un allele nell’esone 10 (ereditata dalla madre)
- la delezione degli esoni 13 e 14 sull’altro allele (ereditata dal
padre).
PIANO DI CURA:
1. Plasmaferesi
2. Terapia farmacologica
andrea.bartuli@opbg.netCurva di risalita del C-LDL dopo LDL-A
1000
900
C-LDLm 237
800
C-LDLm 199
700
C-LDLm 169
600
500
400
300
200
100
0
mg/dl 0 1 2 3 4 5 6 7 10 14 giorni
C-LDLmedio = C-LDLpost + K (C-LDLpre - C-LDLpost)
Kroon formula modified for homozygotes by ThompsonDEPURAZIONE EXTRACORPOREA COSTI - mantenimento accesso venoso - rischi CVC (trombosi, sepsi) - assenza di trials, comparsa di MCV - qualità di vita BENEFICI ritardo comparsa di MCV
FREQUENZA LDL-A 10 giorni 7 giorni C-LDL medio mg/dl 197±30 149±8 FOLLOW-UP di 8 anni (3-11 aa): - Coronarie indenni - Assenza di xantomi - 1 episodio di occlusione del CVC con necessità di rimozione - 1 episodio di sepsi Prognosi: sviluppo di MCV a partire dalla terza decade di vita
Transizione: passaggio dall’età pediatrica a quella adulta Sopravvivenza nell’età adulta modificazione della storia clinica naturale di patologie a mortalità entro l’adolescenza con passaggio all’età adulta in assenza di notizie sulla storia clinica futura
BCM Crisi ricorrenti di addome acuto 1 episodio a 3 anni con appendicectomia 2 successive laparatomie esplorative a 4 anni Comparsa di dolori addominali a domicilio e trasferita da altro PS per pancreatite
Diagnosi di VOLVOLO ricoverata in Pediatria per mancanza posti letto in Chirurgia eseguiva prelievo di routine, ma …….
Dosaggio dei TG 8300
mg/dl, amilasi 2000 UI/l
DIFETTO LPL?
Dosaggio dell’attività LPL: <
3%
Studio genetico: omozigote
Dimessa dopo 15 gg con
dieta ipolipidica
andrea.bartuli@opbg.netFollow up
5-13 anni 1 scompenso ogni due anni circa, TG 725
mg/dl ±220, buon rendimento scolastico; terapia
dietetica, MCT
13-18 anni comparsa di disagio, 1 scompenso ogni 6
mesi circa, TG 1123 mg/dl ± 459, crisi di bulimia seguite
da scompenso acuto, rifiuto dei farmaci, crisi di angoscia,
terapia dietetica, gemfibrozil, LCP, antipertensivi
andrea.bartuli@opbg.net18-25 anni percorso psicoterapeutico, rapporto di coppia stabile……….
andrea.bartuli@opbg.net1: Al-Shali K et al. Successful pregnancy outcome in a patient with severe
chylomicronemia due to compound heterozygosity for mutant lipoprotein lipase.
Clin Biochem. 2002. Mar;35(2):125-30
2: Watts GF et al. Management of patients with severe hypertriglyceridaemia
during pregnancy: report of two cases with familial lipoprotein lipase deficiency.
Br J Obstet Gynaecol. 1992
andrea.bartuli@opbg.netDieta ipolipidica (
Press M et al. Correction of essential fatty-acid deficiency in man by the cutaneous
application of sunflower-seed oil. Lancet. 1974 Apr 6;1(7858):597-8.
- Somministrazione quotidiana transcutanea di olio di semi di girasole sulle braccia e
sul busto (460 mg/die pari a 240 mg di acido linoleico)
andrea.bartuli@opbg.netGravidanza
ricovero se dolori addominali o TG> 3000 mg/dl
Dosaggio emocromo, PCR, Ca, prove emogeniche, amilasi, lipasi, GOT, GPT, γGT
Ecografia addome (pancreatite, litiasi biliare)
NPO con IV glucosata fino a risoluzione dei sintomi
Dieta ipolipidica < 2%
Gemfibrozil dal terzo trimestre 900 mg/die (300 mg tre volte/die)
6 episodi di scompenso con ricovero in Pediatria
andrea.bartuli@opbg.netParto
TC alla 36° settimana, APGAR 1’:9-5’:10, pn Kg 2,810
Allattamento
AL SENO!!!!!
Steiner G, Myher JJ, Kuksis A. Milk and plasma lipid composition in a lactating
patient with type I hyperlipoproteinemia. Am J Clin Nutr. 1985, Jan;41(1):121-8
andrea.bartuli@opbg.netG R A Z I E
Puoi anche leggere

















































