PATOLOGIA GENERALE, CDL IN FARMACIA, III ANNO, 1 SEMESTRE - UNIBA
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Patologia generale, CdL in Farmacia, III anno, 1° semestre
informazioni generali
Programma del corso, testi consigliati, e materiale didattico: https://www.uniba.it/docenti/coluccia-mauro/mauro-coluccia
Testi consigliati:
▪ Robbins Fondamenti di Patologia e di Fisiopatologia, Edra 2013.
▪ Robbins Basic Pathology 10th edition, Elsevier 2018.
Testi sconsigliati: dispense da copisteria (il materiale didattico è relativo ad anni precedenti, gli appunti non sono controllati).
Modalità esame (appelli mensili)
-prova scritta, senza verbalizzazione, con giudizio di idoneità. Tre quesiti a risposta aperta; per l’idoneità alla prova orale è
necessario rispondere adeguatamente ad almeno due quesiti.
-prova orale: esame orale e votazione finale.
Modalità esonero (appello unico, fine novembre-inizio dicembre) (*)
-prova scritta, senza verbalizzazione, con giudizio di idoneità. Tre quesiti a risposta aperta sugli argomenti della prima parte
del programma; per l’idoneità alla prova orale è necessario rispondere adeguatamente a tutti i quesiti.
-prova orale: esame orale sugli argomenti della seconda parte del programma, da sostenersi nei successivi appelli mensili di
gennaio, febbraio o marzo, con votazione finale.
Prof. Mauro Coluccia, Dipartimento di Farmacia (IV piano, stanza 522), tel. 080 5442788, email: mauro.coluccia@uniba.it
Ricevimento: tutte le settimane, martedì e giovedì, h 15.00-17.00 (si consiglia di prenotare l’appuntamento per email).
(*) sospeso per l’AA 2020-2021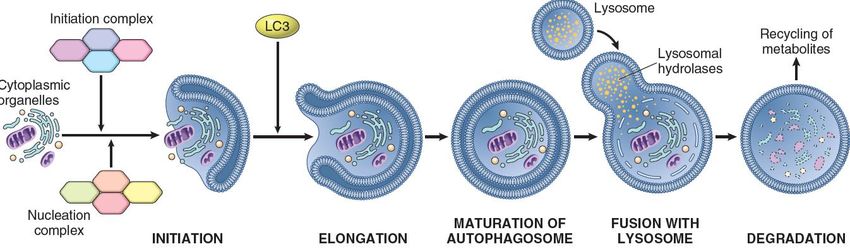
PATOLOGIA: -pathos (emozione profonda, passione, sofferenza, malattia)
-logos (parola, pensiero, principio, discorso, studio).
G.B. Morgagni (1682-1771) R. Virchow (1821-1902) Oggi: Basi cellulari e molecolari dei
«Gli individui sono malati perché le loro processi patologici.
cellule sono malate».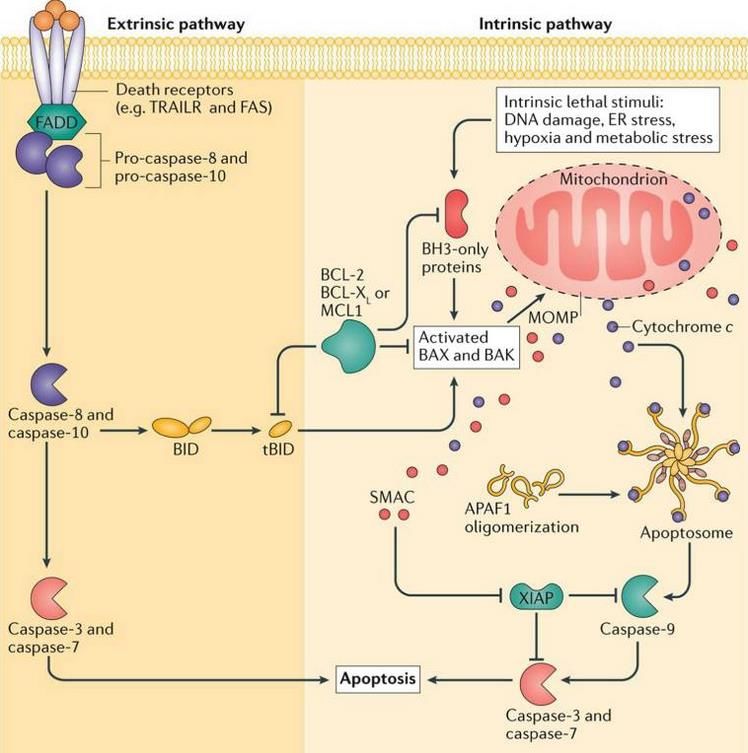
OBIETTIVI FORMATIVI SPECIFICI DEL CORSO DI LAUREA IN FARMACIA
... conferire l’insieme di conoscenze teoriche e pratiche..., alla base di progettazione
strutturale, produzione, regolamentazione, commercializzazione e corretto utilizzo
e controllo del farmaco…
Le discipline teoriche e pratiche del corso di
laurea in Farmacia sono costruite sulla base
della conoscenza dei processi patologici,
vale a dire delle cause delle malattie e delle
conseguenti modificazioni in cellule, tessuti,
organi e apparati del corpo umano...in altri
termini, sulla Patologia generale.
processo patologico
cause danno cellulare/tessutale
▪ infezioni ▪ alterazioni strutturali e
▪ traumi ultrastrutturali
patogenesi
▪ alterazioni nutrizionali ▪ alterazioni funzionali e
▪ alterazioni immunologiche morfologiche di cellule e tessuti
▪ alterazioni genetiche ▪ alterazioni funzionali e
▪ tossine morfologiche e di organi e apparati
manifestazioni cliniche
▪ segni e sintomi di malattia
Quadri principali di risposta cellulare a comuni tipologie di stimoli/eventi lesivi
TIPOLOGIA DELLO STIMOLO/EVENTO LESIVO RISPOSTA CELLULARE
(1) stimoli fisiologici modificati / alcuni stimoli lesivi non letali → adattamento cellulare (vari tipi) *
▪ ↑richiesta funzionale / ↑stimolazione (ad es. ormonale) → ipertrofia e iperplasia
▪ carenza di nutrienti / ridotta stimolazione → ipotrofia - atrofia
▪ stimolo lesivo cronico → metaplasia
(2) eventi lesivi: ad es. ↓apporto di O2 / infezioni / etc. → danno cellulare
▪ evento lesivo moderato e transitorio → danno reversibile
▪ evento lesivo grave e progressivo → danno irreversibile e morte cellulare (necrosi e apoptosi)
(3) alterazioni del metabolismo (genetiche e acquisite) → accumulo cellulare (vari tipi)
(4) evento lesivo sub-letale cronico con effetti cumulativi → invecchiamento cellulare
*NB: in linea di principio, l’adattamento cellulare ha dei limiti, superati i quali si determina una condizione di danno cellulare.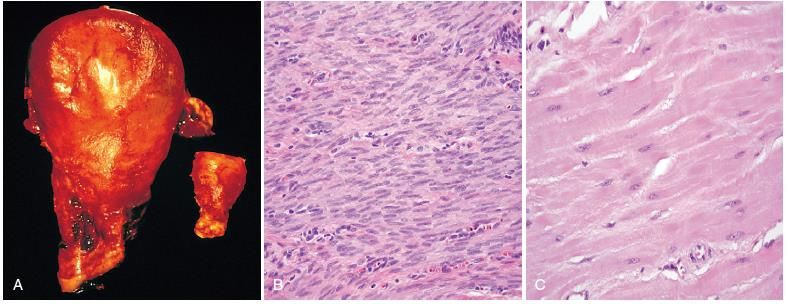
TIPOLOGIA DELLO STIMOLO/EVENTO LESIVO RISPOSTA CELLULARE (1) stimoli fisiologici modificati / alcuni stimoli lesivi non letali → adattamento cellulare (vari tipi) * ▪ ↑richiesta funzionale / ↑stimolazione (ad es. ormonale) → ipertrofia e iperplasia ▪ carenza di nutrienti / ridotta stimolazione → ipotrofia - atrofia ▪ stimolo lesivo cronico → metaplasia
Ipertrofia: aumento delle dimensioni delle cellule (non del numero), e conseguente aumento delle dimensioni dell’organo interessato. (A): dimensioni relative dell’utero normale in gravidanza (sn) e non (dx). (B) cellule muscolari lisce piccole e fusate del miometrio dell’utero non gravido, e (C) grandi cellule muscolari lisce del miometrio in gravidanza (si noti che B e C sono preparazioni osservate a uguale ingrandimento). Meccanismi dell’ipertrofia: azione di segnali molecolari (estrogeni, nel caso specifico), che inducono un’aumentata sintesi di proteine cellulari. Ipertrofia, possibili conseguenze patologiche (esempio dell’Ipertrofia miocardica). L’ipertrofia del miocardio testimonia un adattamento del muscolo cardiaco finalizzato a migliorare la performance meccanica in determinate condizioni (ad es. nell’ipertensione). Tuttavia, l’ingrandimento delle fibre muscolari cardiache ha un limite, superato il quale si produrranno alterazioni cellulari con riduzione della capacità contrattile (quadro clinico dell’insufficienza cardiaca). Si noti come in questo caso, il processo patologico (ipertensione) che inizialmente produce una forma di adattamento cellulare può dar luogo, nella sua evoluzione, a un danno cellulare.

Iperplasia
aumento del numero delle cellule di un tessuto/organo, con conseguente aumento delle dimensioni. Spesso coesiste con
l’ipertrofia, ma è un processo distinto (segnali molecolari che inducono la proliferazione cellulare).
meccanismo esempi
▪ ingrandimento della gh. mammaria nella pubertà e in
Iperplasia fisiologica gravidanza (coesiste con l’ipertrofia)
aumento fisiologico di segnali proliferativi
▪ iperplasia nella rigenerazione epatica
▪ iperplasia del midollo emopoietico (ad es. nelle emorragie)
meccanismo esempi
▪ iperplasia endometriale (alterazione del bilancio estro-
Iperplasia patologica azione eccessiva o inappropriata di segnali progestinico, con relativo aumento di estrogeni e stimolazione
eccessiva della proliferazione delle cellule endometriali).
proliferativi
▪ iperplasia prostatica (da eccessiva stimolazione androgenica)
▪ patologie tumorali
Ipotrofia-atrofia
Riduzione delle dimensioni delle singole cellule di un tessuto/organo, con conseguente riduzione delle dimensioni del
tessuto/organo. Spesso è accompagnata dalla riduzione del numero delle cellule (ipoplasia).
Meccanismo: riduzione della sintesi proteica e/o aumentata degradazione delle proteine.
Atrofia fisiologica Evento normale nello sviluppo embrionale; nell’adulto, involuzione dell’utero dopo il parto.
Riduzione degli appropriati segnali di crescita e proliferazione
▪ da disuso (ad es. atrofia muscolare per prolungata immobilità).
▪ da perdita di innervazione (atrofia muscolare da denervazione)
Atrofia patologica ▪ da riduzione cronica dell’apporto sanguigno (stenosi aterosclerotica)
▪ da malnutrizione
▪ da perdita di stimolazione endocrina
▪ da compressione
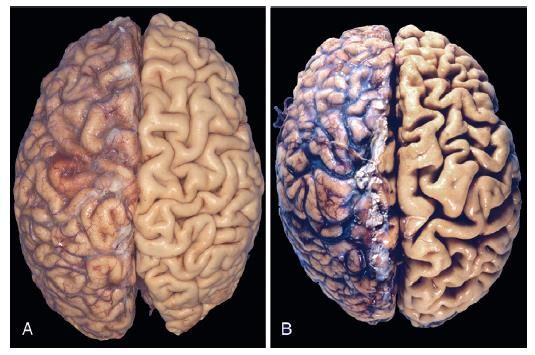
Atrofia cerebrale
(A) Cervello normale, giovane
adulto.
(B) Atrofia cerebrale in 80enne.
Causa: Malattia cerebro-
vascolare su base aterosclerotica.
Si noti come la perdita di cellule
del sistema nervoso riduce le
circonvoluzioni e ingrandisce i
solchi.Metaplasia: modificazione reversibile della differenziazione cellulare
Metaplasia squamosa: esempio di metaplasia in cui l’epitelio cilindrico si
trasforma in epitelio squamoso.
• L’epitelio cilindrico ciliato della mucosa bronchiale si trasforma in
epitelio squamoso pluristratificato a causa dell’esposizione cronica a
stimoli irritativi (ad es. fumo di sigaretta, inquinanti ambientali)
• L’epitelio pluristratificato è più resistente di quello colonnare, ma
questo vantaggio comporta una maggiore suscettibilità alle infezioni
microbiche (perdita della funzione ciliare e riduzione della secrezione di
muco).
Meccanismi: riprogrammazione del programma di differenziazione delle
cellule staminali.
Attenzione, la metaplasia è una
condizione pre-cancerosa: nel
tempo, le cellule metaplastiche
possono diventare neoplastiche.(2) eventi lesivi: ad es. ↓apporto di O2 / infezioni / etc. → danno cellulare ▪ evento lesivo moderato e transitorio → danno reversibile ▪ evento lesivo grave e progressivo → danno irreversibile e morte cellulare (necrosi e apoptosi)
La risposta cellulare agli eventi lesivi: sviluppo progressivo del danno cellulare
cellula normale In condizioni normali, la cellula è capace di mantenere una condizione di stabilità (omeostasi)
(omeostasi) poiché tutti i suoi componenti rispondono in modo integrato alle modificazioni del microambiente.
evento lesivo
In presenza di un evento lesivo, la risposta cellulare sarà finalizzata al raggiungimento di una nuova
danno reversibile condizione di equilibrio e, in relazione alle specifiche circostanze, il danno prodotto potrà essere
reversibile.
severità e persistenza
dell’evento lesivo
danno irreversibile Nel caso di maggiore severità/persistenza dell’evento lesivo, il danno prodotto potrà diventare
e morte cellulare irreversibile e provocare la morte cellulare (con le modalità di necrosi o apoptosi).
▪ In molti comuni eventi lesivi, il danno reversibile si manifesta con il rigonfiamento cellulare (degenerazione idropica o vacuolare): la
cellula e i suoi componenti (in particolare mitocondri e ER) appaiono ingranditi e dilatati; possono essere presenti nel citosol accumuli di
fosfolipidi derivanti da membrane danneggiate (figure mieliniche).Comuni eventi lesivi responsabili di danno cellulare, e loro rilevanza nella pratica medica.
evento lesivo rilevanza
deprivazione di O2 Causa comune e di grande importanza di danno cellulare nella pratica medica (malattie
(ipossia/ischemia) cardiovascolari).
agenti fisici Traumi meccanici, radiazioni, elettricità, variazioni di temperatura e di pressione.
agenti chimici Innumerevoli, possono causare danno sia direttamente che indirettamente.
Dalle rickettsie, ai virus, batteri, e funghi fino ai grandi parassiti: danneggiano le cellule
agenti infettivi con vari meccanismi.
La risposta immunitaria, che normalmente è difensiva, può provocare danno cellulare in
reazioni immunologiche
varie circostanze (immunopatologia).
alterazioni genetiche Alterazioni genetiche ereditarie e acquisite sono alla base di varie malattie.
Comuni cause di danno cellulare: deficit dell’apporto proteico-calorico nei paesi a basso
alterazioni nutrizionali reddito; deficit vitaminici diffusi ovunque; eccessi nutrizionali (ad es. di grassi) nei paesi
ad alto reddito.Il danno cellulare è alla base
della maggior parte dei processi ▪ vi sono innumerevoli e
patologici, ed è un argomento distinti eventi lesivi (cause
molto complesso perché: di danno), di natura diversa
e severità e durata variabili.
▪ nel corpo umano, vi sono alcune centinaia di
tipi e sottotipi cellulari distinti*, e questi
possono mostrare specifiche differenze nella
risposta agli eventi lesivi.
▪ un determinato evento lesivo può avere più
bersagli molecolari nelle cellule che lo
subiscono.
*un «atlante» delle cellule umane è consultabile in https://www.humancellatlas.org/Considerando le tipologie più comuni di evento lesivo, e la loro azione su una cellula
“modello”, il danno cellulare è rappresentato da alterazioni in uno o più componenti
essenziali per il mantenimento dell’omeostasi cellulare/tessutale (1).
Tali componenti possono essere:
• specifiche strutture/organelli (mitocondri, membrane cellulari, DNA
nucleare)
• processi/regolazioni biochimiche le cui alterazioni hanno ripercussioni
generali sulla funzione cellulare (stress ossidativo, omeostasi del Ca2+).
• omeostasi proteica (proteostasi)
1omeostasi (letteralmente «uguale posizione») cellulare, è la capacità della cellula di mantenere le proprie funzioni al variare delle condizioni
dell’ambiente tessutale in cui essa vive.I mitocondri hanno una struttura plastica, controllata da meccanismi di fusione e
fissione, che si adatta al tipo di cellula e alle sue condizioni funzionali.
Funzioni mitocondriali
1. produzione di ATP. 1a. Piruvato (da glucosio ed altri zuccheri) e FA sono trasportati attraverso la IMM nella matrice, dove sono convertiti in
acetil-CoA. L’acetil-CoA entra nel ciclo TCA e viene ossidato a CO2 (che diffonde all’esterno del mitocondrio come prodotto di scarto). L’energia di
legame rilasciata durante l’ossidazione viene in buona parte conservata come elettroni trasportati da NADH. 1b. Il NADH trasferisce gli elettroni
dalla matrice alla ETC della IMM: qui il processo di accoppiamento chemio-osmotico determinerà la trasformazione dell’E trasportata dagli
elettroni nell’E legata al fosfato dell’ATP.
2. ruoli nel metabolismo cellulare.
2.1. regolazione del potenziale redox del citosol. Nella reazione centrale della glicolisi (produzione di difosfoglicerato), la cellula usa NAD+ che diventa
NADH. Poiché il NADH formatosi non può passare la IMM, cede i suoi elettroni a molecole più piccole: queste passano la IMM, cedono elettroni al
NAD+ mitocondriale, e tornano a ricaricarsi nel citosol.
2.2. metabolismo biosintetico del citosol. Per le reazioni di biosintesi sono necessari: ATP (da glicolisi e OxPhos), NADPH (dallo shunt dei pentoso-
fosfati ), e scheletri carboniosi (dalla degradazione degli zuccheri). In condizioni di normale apporto di nutrienti e di ATP, i mitocondri contribuiscono
sia a generare NADPH sia a fornire scheletri carboniosi. Infatti, il citrato generato in eccesso nel TCA va nel citosol, e viene metabolizzato ad acetil-CoA
per la produzione di FA e di steroli.
2.3. ciclo dell’urea. Due step del ciclo dell’urea sono mitocondriali.
2.4. i mitocondri contribuiscono anche alla biosintesi dei gruppi eme e delle membrane, e alla regolazione dell’omeostasi del Ca2+.danno mitocondriale (principali conseguenze)
1. Deplezione di ATP (e conseguenze a valle)
Generalmente causata da ipossia/ischemia, e a volte da agenti tossici, la deplezione di ATP compromette i processi ATP-dipendenti e,
quando la quantità disponibile è il 5-10% del normale, si manifestano più effetti.
1.1. ↓funzione della Na,K-ATPasi, compromissione dell’omeostasi idrico-salina→ rigonfiamento cellulare.
1.2. ↓ATP/ADP → ↑glicogenolisi e glicolisi→ accumulo ac. lattico (e Pi) →↓pH→ ↓riduzione attività di molti enzimi
citosolici.
1.3. Disassemblaggio dell’apparato di sintesi proteica → riduzione della sintesi proteica, steatosi.
2. Aumento delle ROS.
Il danno mitocondriale può provocare una fosforilazione ossidativa incompleta e conseguente aumento delle ROS (discusso più avanti).
3. Interferenza con la funzione mitocondriale nell’omeostasi sopravvivenza-apoptosi (discussa più avanti).
NB: Il danno mitocondriale è spesso testimoniato dalla formazione nella IMM di una proteina canale ad alta conduttanza detta
mitochondrial permeability transition pore (MPTP). MPTP compromette il mantenimento del potenziale di membrana
mitocondriale (↓ATP) e, aumentando la permeabilità a piccole molecole, può determinare ingrandimento degli organelli. Si
consideri che l’apertura di canali mitocondriali interferisce con la funzione degli organelli nell’omeostasi sopravvivenza-apoptosi.danno delle membrane cellulari (caratteristiche e principali conseguenze)
Vari eventi lesivi possono compromettere l’integrità strutturale delle membrane cellulari
1. per ↑degradazione (attivazione di fosfolipasi) e perdita (↑ROS) dei fosfolipidi
2. per ↓sintesi e riacilazione dei fosfolipidi (↓ATP)
3. per danno del citoscheletro (attivazione inappropriata di proteasi).
Compromissione della la funzione di compartimentazione della cellula e dei suoi
componenti.
1. danno della membrana plasmatica: influsso di fluido e soluti,
perdita di componenti intracellulari. NB: Un’aumentata degradazione dei
2. danno delle membrane mitocondriali: apertura MPTP → ↓ATP fosfolipidi e il danno del citoscheletro
(vedi deplezione di ATP, e interferenza omeostasi sopravvivenza- dovuti rispettivamente ad attivazione
apoptosi. inappropriata di fosfolipasi e di
3. danno membrane lisosomiali: rilascio di enzimi e attivazione di proteasi è spesso secondaria ad un
idrolasi che degradano proteine, acidi nucleici e glicogeno. aumento non controllato del Ca2+
citosolico libero.Danno del DNA
Vari agenti lesivi (ROS, radiazioni, agenti
chimici (farmaci citotossici), ed errori
replicativi possono indurre varie
alterazioni strutturali del DNA, che
interferiscono con i normali processi di
trascrizione e replicazione.
La presenza di alterazioni strutturali del DNA è riconosciuta da varie proteine. Questi
sensori attivano vie di segnalazione (DNA damage response, DDR) che agiscono sui
checkpoint della progressione del ciclo cellulare, attivano sistemi enzimatici per la
riparazione del danno, e influenzano l’omeostasi sopravvivenza-morte cellulare.Stress ossidativo Condizione dovuta all’aumentata concentrazione di specie radicaliche (principalmente specie reattive
dell’ossigeno, ROS), non efficacemente controbilanciata dai sistemi antiossidanti. Le alterazioni associate
allo stress ossidativo possono determinare morte cellulare (necrosi o apoptosi, e anche modalità miste:
necroptosi).
Caratteristiche delle specie radicaliche:
• Entità atomiche/molecolari caratterizzate dalla presenza di un elettrone
spaiato, altamente instabili e molto reattive, a vita media brevissima.
Principali specie radicaliche in patologia:
▪ ROS (superossido ●O2-, perossido d'idrogeno H2O2, e
radicale ossidrilico ●OH); perossinitrito ONOO-.
Eventi più comunemente responsabili di stress ossidativo:
▪ Agenti chimici e radiazioni (in particolare radiazioni ionizzanti)
▪ Infiammazione
▪ Danno da riperfusione
▪ Invecchiamento cellulareSpecie reattiva Meccanismi di produzione Meccanismi di Effetti patologici
rimozione
▪ riduzione incompleta di O2 nel
convertito in H2O2 e O2 Danno diretto su
corso della fosforilazione
Superossido (●O2-) dalla SOD (superossido lipidi, proteine e DNA
ossidativa
dismutasi)
▪ ossidasi fagocitica (infiammazione)
convertito in H2O e O2 Può essere
Perossido d’idrogeno (H2O2) Dal superossido per azione di SOD dalla catalasi, e dalla convertito in ●OH e
glutatione perossidasi ipoclorito (OCl-)
Prodotto con varie modalità da H2O, convertito in H2O dalla Danno diretto su
Radicale idrossilico (●OH )
H2O2, e ●O2- glutatione perossidasi lipidi, proteine e DNA
convertito a nitrito da
Interazione fra ●O2- e NO (prodotto Danno diretto su
Perossinitrito (ONOO-) enzimi mitocondriali e
dalla NO sintasi, nell’infiammazione) lipidi, proteine e DNA
citosoliciSistemi antiossidanti
Le specie radicaliche sono intrinsecamente instabili e decadono spontaneamente. Il
decadimento è efficacemente accelerato da sistemi enzimatici e non, ampiamente
distribuiti:
▪ SOD: incrementa significativamente il decadimento del superossido. Nell’uomo
sono presenti tre forme: citoplasmatica, mitocondriale ed extracellulare.
▪ Glutatione (GSH)-perossidasi: enzima citoplasmatico (ma diverse isoforme sono
presenti in altre sedi ad es. nel plasma) che converte H2O2 in H2O e diglutatione
Produzione, rimozione, e ruolo di ROS nel danno cellulare. Molte ossidato (GS-SG). Il calcolo del rapporto intracellulare GSSG/GSH riflette la
circostanze possono determinare un aumento di ROS; l’eccesso di condizione cellulare di stress ossidativo.
produzione e/o l’inadeguato smaltimento determinano un accumulo che ▪ Catalasi: presente nei perossisomi (sede metabolismo ac. grassi a catena molto
può danneggiare lipidi, proteine e DNA. lunga), converte H2O2 in H2O e O2.
SOD, superossidosidmutasi. ▪ Antiossidanti endogeni/esogeni: vit E, vit A, vit C (molecole idrosolubili e
liposolubili).Danno da riperfusione
▪ Danno associato al ripristino della circolazione sanguigna in un tessuto
dopo un periodo di ischemia.
▪ La riperfusione di un tessuto ischemico in cui siano presenti cellule vitali
può produrre un aumento del danno cellulare, principalmente
attribuito a stress ossidativo.
Meccanismi patogenetici
▪ Il danno mitocondriale ostacola il completamento della riduzione progressiva dell’ossigeno molecolare
▪ Possibile compromissione dell’efficacia dei sistemi antiossidanti associata alla condizione ischemica
▪ La riperfusione potenzia la risposta infiammatoria indotta dall’ischemia: i leucociti attivati producono ROS, e
questi danneggiano ulteriormente le cellule in sofferenza ischemica.SOSTANZE TOSSICHE (agenti chimici presenti nell’ambiente, tossine prodotte da agenti infettivi)
Categoria molto variegata di agenti lesivi, determinano un danno cellulare che culmina principalmente nella necrosi.
▪ Sostanze tossiche ad azione diretta: reagiscono direttamente con bersagli cellulari.
▪ Cloruro di mercurio (contaminazione prodotti ittici): alterazione proteine di membrana e inibizione del
trasporto cellulare.
▪ Tossine microbiche: vari tipi di tossine danneggiano specifici componenti cellulari.
▪ Sostanze tossiche ad azione indiretta: l’azione tossica dipende dalla conversione metabolica, da parte di sistemi
microsomiali del RE epatocitario (P450), a intermedi reattivi (spesso specie radicaliche), che danneggiano gli
epatociti (o comunque le cellule con cui vengono a contatto).
▪ Tetracloruro di carbonio (CCl4), acetaminofene (paracetamolo).Alterazioni dell’omeostasi del Ca2+
Il Ca2+ citosolico libero, mediante variazioni controllate della concentrazione, opera da «secondo» messaggero in varie vie di
segnalazione cellulare, implicate nei processi di rimodellamento del citoscheletro (motilità), espressione genica, metabolismo,
esocitosi ed endocitosi, oltre che nella contrazione delle fibre muscolari.
omeostasi del Ca2+
• In condizioni basali, la [Ca2+]c è ∼10-7 M (così come [Ca2+]mt e [Ca2+]n) mentre la [Ca2+]EC è ∼10-3M. Nel reticolo endoplasmatico (e sarcoplasmatico), la [Ca2+]ER è
1→5×10-4M.
• In condizioni basali, il basso livello di [Ca2+]c è mantenuto da PMCA e di NCX, a cui si aggiungono SERCA e (in misura minore) mtCU quando la [Ca2+]c sia
elevata. Tali proteine rilevano e sono attivate dal Ca2+, per cui qualsiasi innalzamento di [Ca2+]c stimola la rimozione del catione, così determinandosi il
controllo omeostatico della [Ca2+]c.
• Vari stimoli (depolarizzazione di membrana, segnali molecolari extracellulari e intracellulari) determinano un aumento di [Ca2+]c > 10-6 M, che può derivare da:
o influsso di Ca2+EC attraverso canali del Ca2+ della membrana plasmatica
o rilascio di Ca2+ER attraverso IP3R e RyR
• L’incremento di [Ca2+]c é generalmente ripido, seguito da una riduzione e da oscillazioni ripetute della [Ca2+]c; queste modificazioni sono supportate da
regolazioni a feedback positivo e negative che sincronizzano l’attività dei canali del Ca2+ e dei meccanismi di rimozione del catione. Ogni citotipo ha una
combinazione unica di canali e pompe del Ca2+ allo scopo di produrre un segnale del Ca2+ citotipo- e agonista-specifico.
[Ca2+]c , [Ca2+]mt, [Ca2+]n, [Ca2+]EC, [Ca2+]ER: concentrazione di Ca2+ a livello citosolico, mitocondriale, nucleare, extracellulare, e del reticolo endo-sarcoplasmatico.
PMCA (plasma membrane Ca2+ transport ATPase), NCX (Na+/Ca2+ exchanger), SERCA (sarcoendoplasmic reticulum Ca2+-ATPase), mtCU (mitochondrial Ca2+ uniporter), IP3R
(1,4,5-triphosphate receptor), RyR (ryanodine receptor).
Essendo l’omeostasi del Ca2+ implicata in processi cellulari fondamentali, un innalzamento incontrollato della [Ca2+]
può determinare danno e morte cellulare.Patogenesi del danno cellulare associato ad alterazioni dell’omeostasi del Ca2+
Ischemia
(e alcuni agenti tossici)
• Alterazioni di pompe ATP-dipendenti
• Squilibrio idrico-salino (Na, K, pH)
↑[Ca2+]c
▪ Attivazione inappropriata di enzimi Ca2+-dipendenti
• fosfolipasi → danno di membrana
• proteasi → danno del citoscheletro → danno di membrana
• endonucleasi → danno del DNA
• ATPasi → ↓ATP (e conseguenze a valle)
▪ ↑[Ca2+]mt → MPTP (e conseguenze a valle, incluso ↓ATP)
NB: in sistemi in vitro, la
sottrazione del Ca2+ protegge dal
danno e morte cellulare danno indotto da svariati eventi
lesivi, ma la rilevanza in vivo dei
meccanismi di danno indicati non
è ancora chiara.Alterazioni dell’omeostasi proteica, proteine mal ripiegate (misfolding)
Proteine
▪ Struttura primaria (conformazione nativa): sequenza lineare degli
aminoacidi che la compongono.
▪ Struttura secondaria: ripiegamenti con formazione di α-elica e
piani β, stabilizzati da ponti H intramolecolari.
▪ Struttura terziaria: ripiegamenti derivanti dall’interazione fra varie
porzioni della catena, che interagiscono tramite le catene laterali
degli aa (legami ionici, ponti H e interazioni idrofobiche). La
struttura può essere ulteriormente stabilizzata da ponti covalenti
S-S. In base alla forma acquisita, le proteine sono classificate come
globulari e fibrose.
▪ Struttura quaternaria: assemblaggio di complessi composti da più
catene polipeptidiche (a struttura terziaria).
Denaturazione delle proteine. Le interazioni molecolari alla base della
struttura secondaria e terziaria (e quaternaria) delle proteine sono
facilmente influenzabili da variazioni di pH e di temperatura (e anche per
azione di raggi UV o di solventi organici), che possono alterare le proteine,
denaturandole. La denaturazione risparmia la struttura primaria ma altera
quella secondaria, terziaria e quaternaria compromettendo le funzioni
biologiche.
Le proteine denaturate coagulano (perdono la solubilità e si aggregano): le
proteine denaturate espongono gruppi che favoriscono la formazione di
legami intramolecolari (sia deboli che forti), che determinano l’aggregazione
di più proteine.folding e misfolding ▪ Le proteine sono strutture “fragili” (interazioni deboli, assoggettate alle condizioni ambientali) e anche in condizioni normali, il processo di ripiegamento produce una certa quantità di misfolding ▪ Nell’ambiente intracellulare, il notevole affollamento molecolare fa sì che una proteina parzialmente ripiegata possa interagire con altre molecule, producendo aggregati potenzialmente nocivi. ▪ Le chaperonine prevengono associazioni inappropriate (con un processo ATP-dipendente, circondano la catena polipeptidica e proteggono il suo processo di ripiegamento fino a quando non è completato). ▪ Le chaperonine provvedono alla “riparazione” delle proteine malripiegate. ▪ Il processo di folding delle proteine sintetizzate a livello del RER avviene all’interno del lume ER, è assistito da chaperonine ed è coadiuvato da reazioni di glicosilazione e di formazione di ponti disolfuro (funzioni facilitate dal Ca2+ER e da un ambiente redox maggiormente spostato verso l’ossidazione rispetto al citosol).
risposta a proteine mal ripiegate (Unfolded Protein Response), e stress reticolo-endoplasmatico
Stimoli fisiologici e patologici Sensori di ER stress
• ↑domanda biosintetica (con e principali vie di
accumulo di proteine e segnalazione della
↑misfolding) UPR
• stress ossidativo
• disomeostasi del Ca2+
• ipossia/ischemia
UPR (unfolded protein response)
• espansione dell’ER • autofagia
ER stress
• regolazione redox • morte cellulare (apoptosi)
• aumento della capacità di folding
• Inibizione della traduzionePrincipali cause di accumulo di ▪ Alterazioni genetiche: mutazioni responsabili di cambiamenti
proteine mal ripiegate della sequenza amminoacidica, con conseguente
modificazione della struttura proteica
▪ Invecchiamento cellulare: progressiva riduzione della capacità
di correggere il mal ripiegamento delle proteine
▪ Infezioni (da microbi intracellulari): aumento della quantità di
proteine da gestire
▪ Modificazioni del pH e dello stato ossidativo intracellulare
Effetti patologici di ▪ Perdita della funzione proteica specifica (mal ripiegamento, e
proteine mal ripiegate (1) aumentata degradazione)
▪ Danno cellulare progressivo da accumulo, esito in apoptosi e
perdita netta di cellule (malattie neurodegenerative)
(1)
Proteine mal ripiegate possono anche accumularsi in sede
extracellulare (ad es. nell’amiloidosi).Esempi di malattie da proteine mal ripiegate
malattia proteina patogenesi
La perdita di CFTR causa difettoso trasporto di Cl e morte
Fibrosi cistica (1) CFTR
cellulare
Ipercolesterolemia familiare (1) Recettore LDL Ipercolesterolemia associata a perdita di LDL-R
Esosaminidasi La perdita dell’enzima lisosomiale determina accumulo
Malattia di Tay-Sachs (1)
(subunità β) neuronale di gangliosidi
Apoptosi dei fotorecettori conseguente a mal ripiegamento
Retinite pigmentosa (2) rodopsina
della rodopsina
Malattia di Creutzfeldt-Jacob (2) prioni Morte neuronale da mal ripiegamento di PrPsc
Malattia di Alzheimer (2) Peptide Aβ Accumulo di amiloide e morte neuronale
Apoptosi epatocitaria da accumulo; enfisema da distruzione
Deficit di α-1 antitripsina (3) α-1 antitripsina
della parete alveolare.
(CFTR, Cystic fibrosis transmembrane conductance regulator; LDL, low-density lipoprotein; PrPsc, Prion protein-scrapie)
Il principale meccanismo patogenetico è rappresentato dall’aumentata degradazione della proteina anomala e alla
conseguente perdita di funzione in (1), all’apoptosi associata allo stress reticolo endoplasmatico in (2), e alla
combinazione di entrambi i meccanismi nel caso (3).Ischemia/Ipossia. L’ischemia (riduzione della perfusione di sangue) è il più comune evento lesivo nella pratica medica.
Il conseguente danno cellulare deriva dalla riduzione dell’apporto alle cellule di O2 (ipossia) e di nutrienti.
▪ Patogenesi del danno ipossico/ischemico
Alterazioni dei processi ATP-dipendenti
▪ trasporto di membrana danno reversibile
Alterazioni del metabolismo energetico ▪ sintesi proteica ↓
• compromissione della OxPhos ▪ lipogenesi danno irreversibile e
▪ turnover fosfolipidico (de-acilazione e ri- morte cellulare (necrosi)
acilazione)
▪ Conseguenze dell’ipossia/ischemia severa-persistente
• ↓attività Na/K ATPasi → accumulo intracellulare di Na e deplezione di K → accumulo di H2O e rigonfiamento cellulare, alterazioni
ioniche (Ca2+).
• ↑ ac. lattico (glicolisi anaerobica), ↓pH intracellulare e ↓attività di vari enzimi cellulari.
• alterazioni strutturali dell’apparato di sintesi proteica (distacco ribosomi dal RER e dissociazione dei polisomi)
• danno irreversibile alle membrane mitocondriali e lisosomiali e alla membrana plasmatica, con esito finale in necrosi.
▪ Risposta cellulare
• attivazione trascrizionale di HIF-1 (hypoxia-inducible factor 1)
• ↑VEGF (vascular endothelial growth factor) e stimolazione dell’angiogenesi
• Modificazioni del metabolismo energetico (↑captazione di glucosio e glicolisi anaerobica, ↓fosforilazione ossidativa)Il fattore di trascrizione HIF-1 e la risposta adattativa all’ipossia
E3 ligasi
VHL
PHD2
OH-Pro
HIF-1
OH-Asn
FIH-1
p300 proteasoma
In condizioni di ossigenazione normale, HIF-1 è mantenuto inattivo... ...ma, in condizioni di ipossia, HIF1 è attivato
▪ l’enzima PHD2 (prolil idrossilasi dominio 2) usando l’ossigeno
▪ le reazioni di PHD2 e FIH-1 sono inibite, HIF-1 si accumula, dimerizza, recluta p300,
idrossila un residuo di prolina di HIF-1. In tale forma, HIF-1 recluta
e quindi attiva la trascrizione di centinaia di geni bersaglio.
la proteina VHL (von Hippel Lindau) che a sua volta recluta una E3
▪ il programma trascrizionale HIF-1 è finalizzato a:
ligasi (ubiquitina ligasi). Questa, ubiquitinando HIF-1 lo avvia alla
▪ aumentare la fornitura di O2 (EPO, eritropoietina)
degradazione proteasomica.
▪ promuovere l’angiogenesi (VEGF)
▪ L’enzima FIH-1 (fattore di inibizione di HIF-1) usando l’ossigeno ▪ modificare il metabolismo energetico (PDK1, piruvato deidrogenasi chinasi
idrossila un residuo di asparagina di HIF-1. In tale forma, HIF-1 1, per l’inibizione del ciclo TCA; aumento GLUT1 ed enzimi glicolitici).
non può reclutare la proteina co-attivatrice p300, essenziale per
la sua funzione trascrizionale.Regolazione del metabolismo energetico (del glucosio) in condizioni di normossia e ipossia.
Normossia. Gli enzimi glicolitici convertono il
glucosio in piruvato. La piruvato deidrogenasi
(PDH) converte il piruvato in acetil-CoA. AcCoA si
ossida nel ciclo TCA e genera elettroni che
vengono trasportati attraverso una serie di
complessi proteici (catena di trasporto
elettronica, ETC) e infine trasferiti sull’ossigeno
per formare acqua. Il trasporto di elettroni lungo
la ETC genera il gradiente protonico utilizzato per
la sintesi di ATP.
Ipossia. La piruvato deidrogenasi chinasi (PDK1)
inibisce PDH; la lattato deidrogenasi (LDHA)
converte il piruvato in lattato. Inoltre, in
condizioni di ipossia, la captazione di glucosio
(GLUT1) e l’espressione degli enzimi glicolitici
aumentano (non indicato in figura).Danno persistente/severo ed esito finale in necrosi ▪ Le conseguenze morfologiche e funzionali dell’ipossia/ischemia dipendono dalla gravità e dalla durata dell’evento lesivo. ▪ Il confine fra condizione reversibile e irreversibile è di difficile individuazione. ▪ L’irreversibilità è testimoniata dalla distruzione strutturale delle membrane mitocondriali e lisosomiali e infine dalla distruzione della membrana plasmatica con fuoriuscita dei componenti intracellulari e induzione/incremento della risposta infiammatoria.
Rene: danno reversibile e irreversibile (necrosi). (A) epitelio tubulare normale. (B) epitelio tubulare con danno cellulare reversibile (architettura conservata, cellule rigonfie ma ben distinguibili). (C) epitelio tubulare con necrosi (cellule uniformemente eosinofile, nuclei generalmente indistinguibili, architettura scarsamente conservata.
Necrosi
cambiamento morfologico del tessuto, conseguente alla morte cellulare. L’aspetto morfologico macroscopico e
microscopico della necrosi deriva dalla 1) denaturazione delle proteine cellulari, e dalla 2) digestione enzimatica dei
costituenti delle cellule morte.
Forme principali
NECROSI COAGULATIVA: tipica conseguenza di ischemia/ipossia in tutti gli organi con l’eccezione del cervello. Prevalgono i
fenomeni di denaturazione/coagulazione delle proteine, e l’architettura del tessuto è mantenuta per giorni dopo la morte
cellulare. Micro: materiale uniforme, eosinofilo; i contorni cellulari sono distinguibili, scompaiono i nuclei.
NECROSI COLLIQUATIVA: tipica conseguenza di infezioni (tutti gli organi) e dell’ischemia cerebrale. Legata al rilascio di
enzimi idrolitici (da batteri e da leucociti neutrofili). Macro: il tessuto è liquefatto (a volte giallastro per la presenza di
pus). Micro: evidente infiltrato infiammatorio, soprattutto leucociti neutrofili.
Varianti
Necrosi caseosa: tipica necrosi tubercolare. Macro: tessuto biancastro (simile al formaggio). Micro: parte centrale uniformemente
eosinofila circondata da linfociti e macrofagi attivati (cellule giganti, e cellule epiteliodi). Nell’insieme, la struttura è denominata granuloma
tubercolare.
Steatonecrosi (necrosi grassa): necrosi di tessuti con ricca componente adipocitaria (ad es pancreas, mammella). Le cellule morte rilasciano
enzimi litici che degradano i lipidi formando acidi grassi liberi. Macro: depositi biancastri per formazione di saponi di Ca. Micro: adipociti
anucleati con depositi di calcio.
Necrosi fibrinoide: osservabile in caso di danno vascolare. Macro: nessuna particolarità. Micro: depositi di fibrina all’interno dei vasi.
Necrosi gangrenosa: termine clinico per necrosi ischemica degli arti inferiori (a volte superiori). Macro: colorito nerastro con vari gradi di
putrefazione. Micro: combinazione di necrosi coagulativa (ischemia) e colliquativa (gangrena umida per sovrainfezione batterica).La morte cellulare
necrosi apoptosi
• Morte cellulare «accidentale», caratterizzata da • Negli organismi complessi, la morte cellulare è un
rigonfiamento cellulare e rottura delle evento molto comune che, nella maggior parte
membrane, distruzione degli organelli, il più delle dei casi, avviene con una modalità chiamata
volte senza condensazione della cromatina. apoptosi (*).
• La necrosi consegue generalmente a un danno • L’ apoptosi, o morte cellulare programmata, è
massivo e irreparabile di componenti e funzioni caratterizzata da contrazione cellulare, e
cellulari fondamentali per l’omeostasi. A questa condensazione della cromatina. Il processo è
forma «classica» di necrosi, si è aggiunta una attivato da specifiche vie di segnalazione
modalità regolata da segnali molecolari (necrosi cellulare (intrinseca ed estrinseca), ed è eseguito
programmata, o necroptosi). da specifiche cistein-proteasi (caspasi).
autofagia
• Modalità di morte caratterizzata dalla presenza di grandi vescicole intracellulari (autofagosomi) contenenti ampie
porzioni di citoplasma e organelli.
• Meccanismo di sopravvivenza che risponde a crisi metaboliche o rimuove organelli danneggiati. Qualora il meccanismo
non sia sufficiente, si determina morte cellulare accompagnata da autofagia.
* Il corpo umano comprende 3.72 × 1013 cellule: di queste, si calcola che 50 x 109 muoiano ogni giorno per apoptosi.▪ Processo di morte cellulare mediante il quale definite
cellule vengono eliminate dal sistema/tessuto al quale
appartengono.
▪ Come risultato, si determina un controllo del numero e
del tipo di cellule che compongono un determinato
sistema.
▪ L’apoptosi (diversamente dalla necrosi) interessa quindi
«singole» cellule, e non induce una reazione
infiammatoria.
Apoptosi: caratteristiche morfologiche▪ «morte cellulare programmata», durante lo sviluppo
embrionario e fetale.
▪ involuzione tessutale ormono-dipendente (ad es.
regressione endometriale post-menopausale, regressione
Apoptosi in condizioni fisiologiche mammaria dopo lo svezzamento).
▪ regolazione della risposta immunitaria (ad es. eliminazione
dei linfociti autoreattivi).
▪ controllo qualitativo e quantitativo delle cellule in
popolazioni proliferanti (ad es. tessuto epiteliale ed
emopoietico).
▪ svariati stimoli dannosi possono determinare apoptosi
Apoptosi in condizioni patologiche (piuttosto che necrosi), in relazione allo stimolo (tipo,
durata, entità), e al tipo e alle condizioni funzionali delle
cellule interessate.L’apoptosi è una sorta di «smontaggio» ordinato delle varie strutture e componenti cellulari. É eseguito da enzimi
specializzati chiamati caspasi (cistein proteasi) che, in una regolazione a cascata, danno luogo alla produzione dei corpi
apoptotici, che verranno poi internalizzati dai fagociti.
Le caspasi sono raggruppate in due tipi:
▪ caspasi iniziatrici (2, 8, e 9), formate da un prodominio,
una subunità grande ed una subunità piccola.
▪ caspasi esecutrici (3, 6 e 7), formate da una subunità
grande e da una piccola.
Le procaspasi iniziatrici, attraverso i loro prodomini,
vengono reclutate su “piattaforme di attivazione” (non
mostrate) dove dimerizzano e si attivano (attivazione per
prossimità) in caspasi.
Le caspasi iniziatrici le procaspasi esecutrici operando un
taglio fra la subunità grande e quella piccola.
Le caspasi esecutrici tagliano centinaia di substrati.
NB: le caspasi cataliticamente attive sono tetrameri (due
subunità grandi e due piccole).Schema generale dell’apoptosi
• L’attivazione delle caspasi iniziatrici avviene con due distinte
modalità, estrinseca ed intrinseca. Una volta attivate, le
caspasi iniziatrici (caspasi 8 e 10 sulla via estrinseca e caspasi
9 sulla via intrinseca) attivano le caspasi esecutrici 3, 6 e 7.
• Nella via estrinseca, molecole di pro-caspasi 8 (o 10) sono
reclutate a livello di recettori di morte cellulare attivati dai
rispettivi ligandi (ad es. TRAIL-TRAILR, FASL-FAS), su cui hanno
interagito proteine adattatrici (FADD). Il reclutamento delle
pro-caspasi ne favorisce la dimerizzazione e l’attivazione per
prossimità.
• Nella via intrinseca (o mitocondriale), l’attivazione della pro-
caspasi 9 avviene su una piattaforma molecolare detta
apoptosoma, per la cui formazione è necessaria la
permeabilizzazione della membrana mitocondriale esterna
(MOMP). Con la MOMP, Cyt c e SMAC (e OMI) sono rilasciati
nel citosol. Cyt c, interagendo con APAF1, determina
l’assemblaggio dell’apoptosoma. La procaspasi 9 viene
continuamente reclutata sull’apoptosoma, attivata per
prossimità, e rilasciata come caspasi 9. SMAC (e OMI)
facilitano l’apoptosi inibendo XIAP.
• La condizione MOMP dipende dall’azione delle proteine BAX e
BAK attivate, per mezzo di proteine BH3-only, in svariate
condizioni di danno cellulare.
Si noti che le caspasi 8 e 10 (via estrinseca) possono attivare
le proteine BAX e BAK, avviando in tal modo l’attivazione
della via intrinseca.
Figura tratta da: S.H. Suhaili et al. doi: 10.1007/s12551-017-0308-0Apoptosi, via intrinseca (mitocondriale), ruolo delle proteine bcl-2.
• Le proteine della famiglia bcl-2 (sono caratterizzate da
regioni conservate (domini BH), e classificate come multi-
dominio e BH3-only.
• Le proteine della famiglia bcl-2 sono distinte funzionalmente
in anti-apoptotiche (bcl-2, bcl-xL...), pro-apoptotiche (Bax,
Bak, Bok), e BH3-only con funzione di attivazione diretta (Bid,
Bim, Puma) o di antagonismo (de-repressione) (Bad, Bik...)
• L’attivazione di Bax/Bak è innescata da un
legame transitorio di BH3-only (Bid e
Bim, attivazione diretta), oppure da
un’interazione con antagonismo delle
• Le proteine pro-apoptotiche sono presenti nella cellula in forma inattiva, nel proteine bcl-2 antiapoptotiche.
cytosol (Bax), o associate alla OMM (Bak).
• Quando vengono attivate, Bax e Bak omo-oligomerizzano e si inseriscono nella
OMM formando pori (MOMP).
• Le proteine anti-apoptotiche localizzate sia a livello mitocondriale sia a livello
del RE possono sequestrare le forme attivate monomeriche di Bax e Bak (non
mostrato in figura).Apoptosi, via intrinseca (mitocondriale): principali circostanze di attivazione, meccanismi e implicazioni.
Stimolo apoptotico Meccanismo di attivazione Implicazioni / esempi
In presenza di danno sul DNA da agenti genotossici
▪ attivazione di p53 e aumento della trascrizione di bcl2 pro- (o da difetto dei sistemi di riparazione), l’apoptosi
Danno del DNA apoptotiche scongiura l’introduzione di alterazioni genetiche
▪ attivazione della degradazione di bcl2 antiapoptotiche nelle cellule figlie (e quindi un aumento del tasso
mutazionale).
▪ nello sviluppo del SN, eliminazione di precursori
neuronali che non riescono a migrare o
innervare il target in modo appropriato.
▪ nella risposta immunitaria, eliminazione dei
Riduzione di segnali di ▪ perdita di funzione della via PI3K/Akt linfociti attivati per deprivazione di citochine
sopravvivenza- crescita ▪ attivazione della degradazione di bcl2 antiapoptotiche (NB: anche coinvolta la via estrinseca).
▪ anoikis: quando cellule epiteliali o endoteliali si
staccano dalla ECM, le integrine perdono il
contatto con recettori ECM e cessano di
influenzare vie di sopravvivenza.
Accumulo di proteine malripiegate a livello di ER, attivazione di L’apoptosi è attivata se i meccanismi di
Stress reticolo-
sensori, attivazione della trascrizione di proteine bcl2 degradazione non riescono a gestire l’accumulo
endoplasmico (ER stress)
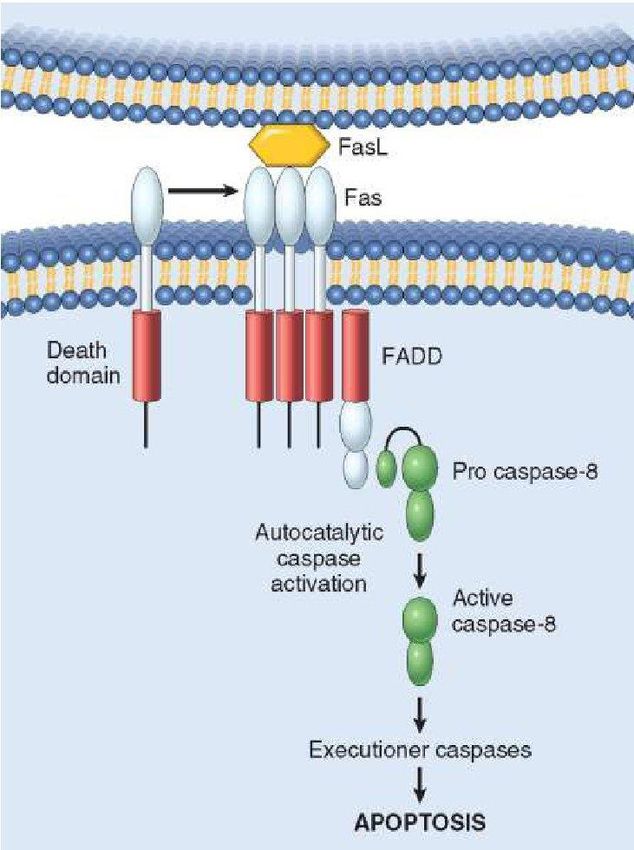
proapoptotiche. proteicoApoptosi, via estrinseca (death receptor, DR), caspasi iniziatrice: CASP-8.
Attivata dall’interazione fra segnali di morte (FasL, TRAIL, TNF) e corrispondenti recettori Fas, TRAILR, TNFR
▪ Quando Fas (o TRAILR) sono ingaggiati dai rispettivi ligandi FasL (o TRAIL) si
determina un raggruppamento e il successivo reclutamento di proteine
adattatrici FADD, formando così la piattaforma di attivazione DISC (death
inducing signaling complex).
▪ DISC recluta pro-caspasi 8 determinandone l’attivazione per prossimità. In
alcune circostanze, casp 8 taglia e attiva le casp esecutrici 3 e 7. In altre
circostanze, in cui XIAP inibisce le caspasi esecutrici, la casp 8 taglia la BH3-only
bid, la quale attiva Bax/Bak e MOMP (via mitocondriale). Il rilascio di Smac e
Omi inibisce XIAP (non mostrato).
▪ L’interazione TNF-TNFR attrae TRADD, e avvia una segnalazione più complessa
con tre distinti possibili esiti dipendenti dalla condizione funzionale delle
proteine reclutate sulla piattaforma: sopravvivenza e risposta infiammatoria,
apoptosi, necrosi programmata (necroptosi) (non mostrato).
NB: La via estrinseca è un meccanismo fondamentale nella risposta immunitaria.Processi patologici associati ad alterata regolazione dell’apoptosi
▪ la sopravvivenza di cellule con danni al genoma è un importante meccanismo di
cancerogenesi ed è una caratteristica fondamentale delle cellule tumorali.
difetto dell’apoptosi
(↑sopravvivenza cellulare)
▪ la sopravvivenza di cellule potenzialmente nocive (ad es. linfociti autoreattivi) è un
importante meccanismo delle malattie autoimmunitarie.
▪ nelle malattie neurodegenerative, si ha una perdita di specifiche popolazioni di
eccesso di apoptosi neuroni (apoptosi per UPR).
(↑ mortalità cellulare)
▪ morte di cellule infettate da virusProcessi del catabolismo intracellulare: la degradazione lisosomiale. Nella eterofagia (a Dx), i lisosomi si fondono con endosomi o fagosomi e degradano il materiale contenuto all’interno. I prodotti così ottenuti possono essere rilasciati nel citosol (metabolismo cellulare), o rilasciati nello spazio extracellulare (esocitosi). Nell’autofagia (a Sn), organelli senescenti/danneggiati e aggregati proteice sono avviati alla degradazione mediata da lisosomi dopo essere stati racchiusi da una membrana a doppio strato derivata dal RE e marcata con proteine LC3.
Autofagia Autofagia. Condizioni di stress cellulare (ad es. la deprivazione di nutrienti) attivano il processo dell’autofagia, in cui è possibile distinguere più fasi: iniziazione, nucleazione ed elongazione della membrana isolante → produzione di vacuoli a doppia membrana, autofagosomi, che sequestrano organelli e altro materiale citoplasmatico → dopo la fusione con lisosomi, si formano gli autolisosomi, in cui il materiale viene degradato da idrolasi lisosomiali e reso disponibile per il riciclaggio di metaboliti.
ruolo patologico dell’autofagia
tumori ▪ l’autofagia potrebbe rappresentare un meccanismo di crescita delle cellule tumorali
▪ in alcune malattie neurodegenerative, l’autofagia risulta accelerata e i difetti
malattie neurodegenerative
dell’autofagia accelerano la neurodegenerazione (modelli genetici).
▪ alcuni batteri (ad es. micobatteri, shigella) sono degradati mediante autofagia. Le
malattie infettive alterazioni dei meccanismi di autofagia possono determinare aumentata
suscettibilità a contrarre la TBC.Patologie cellulari da accumulo
Quantità anomale di varie sostanze possono accumularsi nel citoplasma, in organelli (soprattutto lisosomi), o nel nucleo
producendo danno cellulare.
Meccanismi/tipologie di accumuloPatologie da accumulo
Invecchiamento cellulare progressivo declino della vitalità e delle funzioni NB: L’invecchiamento dell’organismo riflette
cellulari associato a (i) alterazioni genetiche e al l’invecchiamento delle cellule che lo compongono
(ii) progressivo accumulo di danno cellulare
dovuto alle condizioni ambientali.
Bersagli biologici e meccanismi dell’invecchiamento cellulare
▪ le alterazioni mutazionali che si accumulano nel tempo, a seguito dell’esposizione ad
agenti genotossici, potrebbero avere un ruolo importante.
Danno del DNA, integrità genomica ▪ alterazioni ereditarie che riguardano enzimi coinvolti nelle funzioni del DNA (ad es.
DNA elicasi nella sindrome di Werner, e in quella di Bloom) si manifestano (anche)
con invecchiamento precoce.
▪ arresto replicativo legato al progressivo accorciamento dei telomeri
Senescenza cellulare
▪ aumentata espressione di geni che codificano per inibitori di chinasi ciclino-
(o senescenza replicativa)
dipendenti.
▪ alterazioni dei processi legati al “folding” delle proteine (chaperonine), e alla
Omeostasi proteica
degradazione di proteine mal ripiegate (autofagia, sistema proteasomico).
▪ via dell’insulina e del fattore IGF-1 (insulin-like growth factor 1): l’attivazione
Deregolazione di vie di segnalazione
promuove la crescita e la proliferazione cellulare.
influenzate dalla condizione nutrizionale (la
▪ sirtuine: deacetilasi NAD-dipendenti, promuovono l’espressione di geni che
restrizione calorica allunga la vita).
influenzano la riparazione del DNA e l’omeostasi proteica.• Il danno del DNA, la senescenza cellulare e le alterazioni dell’omeostasi proteica sono i più noti meccanismi che possono accelerare l’invecchiamento cellulare. • Al contrario, alcune vie di segnalazione influenzate dalla condizione nutrizionale (principalmente la restrizione calorica) rallentano l’invecchiamento cellulare.
Ruolo dei telomeri e della telomerasi nella senescenza replicativa
(A) La successione delle divisioni cellulari associata
all’età determina un progressivo accorciamento
dei telomeri, così innescando il programma di
senescenza e la contrazione dei pool delle
cellule staminali.
(B) L’accorciamento dei telomeri è più marcato
nelle cellule somatiche (di regola non esprimono
la telomerasi), mentre le cellule staminali
(telomerasi-positive) sono capaci di più cicli
replicativi.
Le cellule tumorali spesso attivano la telomerasi e
mantengono indefinitamente la capacità
replicativa.Puoi anche leggere

















































