Capitolo 2 - EZIOLOGIA, PATOGENESI E DIAGNOSI DELLA BSE
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Capitolo 2 - EZIOLOGIA, PATOGENESI E DIAGNOSI DELLA BSE
Le malattie cosiddette “da prioni” o encefalopatie spongiformi trasmissibili sono una
categoria di processi morbosi neurodegenerativi che colpiscono sia l’uomo sia gli animali. Si
parla di forme trasmissibili perché possono essere trasmesse ai mammiferi attraverso
l’inoculazione di tessuti infetti o, in determinati casi, attraverso la via alimentare. Queste
malattie sono tutte associate all’accumulo negli encefali colpiti di una glicoproteina abnorme
isoforma codificata dall’ospite, la proteina prionica (PrP), che sembra essere la componente
principale, forse essenziale (probabilmente la sola), dell’agente trasmissibile o prione. La
PrPsc(1) isoforma, correlata alla malattia, può essere differenziata da quella cellulare normale
isoforma PrPc(2) per la sua insolubilità e per la parziale resistenza alle proteasi. La PrPsc
deriverebbe dalla PrPc per un meccanismo post traslazionale che sembra implicare una
modificazione di conformazione piuttosto che covalente.
Le ricerche di genetica molecolare umana a livello transgenico e gli studi di
conversione in vitro propongono un modello di propagazione dei prioni che comprende
un’interazione diretta proteina-proteina tra la PrPc dell’ospite e la PrPsc inoculata; la PrPsc
agisce in modo tale da promuovere un’ulteriore conversione della PrPc in PrPsc tramite un
processo autocatalitico che procede a cascata in modo più efficiente quando le proteine
interagenti presentano la stessa struttura primaria.
Oltre alla biologia unica e peculiare di queste malattie, che ha sempre fortemente
richiamato l’attenzione di moltissimi ricercatori, la comparsa e lo sviluppo in forma
epidemica della BSE in Gran Bretagna e successivamente anche in altri Paesi Europei, ha
incrementato ancor più l’interesse degli studiosi e dell’opinione pubblica per l’eventualità che
le TSE rappresentino una seria minaccia per la salute pubblica, soprattutto a seguito
dell’ingestione di carni infette di bovini colpiti dalla BSE. Tra l’altro la BSE è anche
responsabile di TSE in un certo numero di altre specie animali come i gatti domestici (FSE),
ungulati esotici in cattività (Nyala e Kudu), presumibilmente a seguito dell’ingestione di carni
contaminate dall’agente dell’encefalopatia bovina.
Nell’uomo esistono diversi tipi di encefalopatie spongiformi che sostanzialmente
possono essere distinte in ereditarie, acquisite e sporadiche. All’incirca il 15% di queste forme
è costituito da malattie ereditarie associate a mutazioni della codificazione nel gene della
proteina prionica (PRNP). Le malattie acquisite comprendono il Kuru e la CJD iatrogena. Vie
iatrogene di trasmissione ben note sono i trattamenti con ormone della crescita o
gonadotropina ricavati dall’ipofisi di cadaveri, i trapianti di dura madre o di cornea, e l’uso di
strumenti neurochirurgici non adeguatamente sterilizzati. Comunque la maggior parte delle
encefalopatie spongiformi umane si manifesta come CJD sporadica, nella quale o per la quale
mancano i riferimenti relativi a mutazioni patogene della PRNP o una storia di esposizione
iatrogena.
La comparsa di una nuova forma di encefalopatia registrata soprattutto in Gran
Bretagna, che colpisce inaspettatamente giovani persone e presenta aspetti clinico-patologici
del tutto caratteristici, fa ritenere che ci si trova di fronte ad una nuova entità nosologica con
ogni probabilità diversa dalla CJD sporadica, e per questo denominata ”nuova variante CJD”,
che potrebbe essere attribuita all’esposizione a carni (o meglio frattaglie) di bovini infettati
dall’agente della BSE.
Tutti i pazienti affetti dalla vCJD e sottoposti ad esame neuropatologico e di
laboratorio (immunoistochimica, immunoblot, ecc.) sono omozigoti per la metionina al
residuo polimorfo 129 della PrP e non offrono a considerare alcuna mutazione della
42 _______________________________________________________________________________________
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001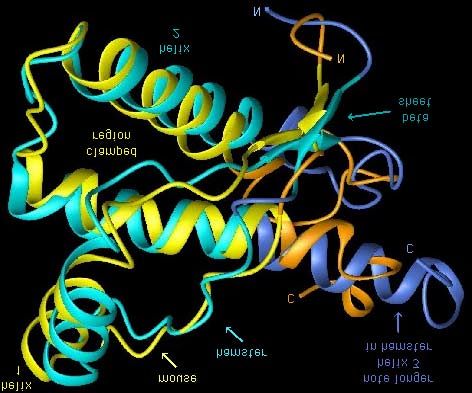
CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
codificazione. Inoltre nessuno di questi ha una storia di esposizione iatrogena a prioni umani.
Studi condotti con la tecnica del western blotting hanno accertato che il tracciato
della PrP resistente alla proteasi, determinato dalla minore o maggiore glicosilazione della
proteina è identico per i casi di vCJD mentre è diverso per la forma classica sporadica e per la
forma iatrogena della malattia.
Poiché le caratteristiche dei tracciati (spessore ed intensità delle bande) di PrP
proteasi resistenti della vCJD differiscono sensibilmente da quelle della CJD sporadica e
iatrogena, si è verificato se il modello distintivo di glicosilazione si potesse rilevare anche
nella BSE, trasmessa naturalmente o sperimentalmente. La forma glicosilata della vCJD è
stata così rilevata nella stessa BSE, nella BSE sperimentalmente trasmessa al topo e nella
BSE trasmessa sperimentalmente al gatto domestico e sempre sperimentalmente al macaco,
avvalorando l’ipotesi che la vCJD origini dalla trasmissione della BSE all’uomo. Secondo
Ironside (1998) tuttavia questo particolare tipo di PrP altamente glicosilata è stato identificato
anche in un ceppo di scrapie e di insonnia fatale familiare e non è pertanto specifico per BSE
e vCJD.
L’eziologia della CJD sporadica o classica (forma cosiddetta spontanea) resta non
definita: al suo determinismo potrebbero concorrere o contribuire mutazioni somatiche della
PRNP o una conversione spontanea della PrPc in PrPsc come raro evento stocastico. La
nuova variante CJD offre d’altro canto a considerare caratteristiche di ceppo nettamente
distinte rispetto agli altri tipi di CJD, e simili a quelle della BSE, lasciando aperta l’ipotesi di
trasmissione bovino-uomo.
(1) sc = scrapie
(2) c = cellulare
_______________________________________________________________________________________ 43
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Le encefalopatie spongiformi
Le encefalopatie spongiformi sono classificate fra le malattie da agenti non
convenzionali. Sono malattie neurodegenerative con esito sempre fatale, colpiscono sia
l'uomo sia gli animali e possono manifestarsi come malattie sporadiche, genetiche od
infettive.
Se ne fornisce un quadro riassuntivo distinguendo quelle che colpiscono i ruminanti,
animali diversi dai ruminanti e l’uomo.
RUMINANTI (Malattie scrapie simili) (modif. da Prusiner, 1997)
• Scrapie
La scrapie è una malattia endemica degli ovini e dei caprini chiamata così perché causa
inizialmente prurito (to scrape: grattare); ciò porta gli animali colpiti a grattarsi ed a
mordersi sino all'autolesionismo. È una patologia infettiva che può essere trasmessa per
via orale con l'ingestione di materiale organico infetto, per via orizzontale
(principalmente nel periodo dei parti) e, probabilmente, per via materno-fetale.
L'insorgenza della malattia è influenzata dal ceppo infettante coinvolto e dalla
sensibilità dell'individuo. Tale sensibilità deriva da profili genetici differenti presenti nei
singoli ovini.
• Encefalopatia spongiforme del bovino (BSE)
L'encefalopatia spongiforme bovina (BSE) è una malattia infettiva fatale del bovino,
comparsa nel 1986 in Gran Bretagna. La prima diagnosi ufficiale fu effettuata
nell'ottobre dello stesso anno. I principali sintomi caratteristici osservati sono
ipersensibilità agli stimoli esterni e incoordinazione degli arti posteriori, con spiccate
reazioni di timore. È probabile che sia un'infezione alimentare, trasmessa tramite farine
animali trattate in maniera insufficiente ad eliminare l'agente eziologico.
• Encefalopatia degli ungulati esotici
È una patologia infettiva assimilabile alla scrapie che è stata osservata principalmente in
ungulati selvatici (orice, antilope, nyala, kudu ecc.) allevati in cattività ed alimentati con
mangimi contaminati.
• Sindrome del dimagrimento cronico
Chronic wasting disease (CWD) è una patologia cronica di origine oscura, simile alla
scrapie, che è stata evidenziata nell'alce delle Montagne Rocciose e nel cervo-mulo
tenuto in cattività o allo stato libero.
44 _______________________________________________________________________________________
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
NON RUMINANTI
• Encefalopatia trasmissibile del visone (TME)
La TME è una malattia che colpisce tipicamente i visoni allevati; è probabile che la
causa sia da attribuire all’alimentazione, inizialmente per il consumo di farine di carne
di animali infetti da scrapie, e successivamente a partire dalle carcasse degli stessi
visoni infetti.
L’agente causale possiede le stesse caratteristiche fisiche, chimiche e biologiche della
scrapie, non è correlato alla BSE e non è trasmissibile, per via naturale, a specie diverse
dal visone.
Il visone si infetta alimentandosi con mangimi contaminati dall’agente causale della
scrapie o direttamente attraverso soluzioni di continuo della cute o delle mucose.
L’incubazione è di 8-9 mesi. La malattia si manifesta con incoordinazione dei
movimenti, cambiamenti nelle abitudini alimentari e minor cura nella toelettatura;
inoltre sono frequenti sintomi di eccitazione. La morte sopraggiunge in 3-8 settimane.
• Encefalopatia spongiforme dei felini (FSE)
La FSE è una malattia osservata nei gatti ed in alcuni altri membri della famiglia dei
felini come gli ocelot, i ghepardi ed i puma ecc. Si è ipotizzato che l'insorgenza di tale
encefalopatia spongiforme sia dovuta all'alimentazione dei felini (serragli, zoo ecc.) in
cattività con materiale infetto da BSE.
La FSE ha un certo riscontro nel Regno Unito, dove è stata diagnosticata la prima volta
nel 1990 ed ha fatto registrare 85 casi dal 1990 al mese di giugno 1999.
Nel nostro paese è conosciuta una forma di encefalopatia spongiforme felina, ma
mancano dati statistici precisi e comprovati da diagnosi di laboratorio che la attribuisca
a FSE.
Il primo sintomo dell’encefalopatia spongiforme felina è rappresentato da mutamenti
nel comportamento, quali aggressività ingiustificata o tendenza a nascondersi. L’atassia
locomotoria è costante; al contrario appaiono solo talvolta i seguenti sintomi: iperestesia
a stimoli uditivi o tattili, polifagia, grooming esagerato o ridotto, ipersalivazione,
polidpsia, midriasi, tremori.
_______________________________________________________________________________________ 45
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
UOMO
• Kuru
Malattia scoperta da Gajdusek nella tribù Fore della Nuova Guinea (1967),
caratterizzata da lungo periodo di incubazione, ipereccitabilità, tremori, atassia, riso
incontrollato, detta anche "morte che ride" per la discinesia facciale. È di origine
alimentare legata al cannibalismo rituale: la tribù onorava i defunti mangiandone il
cervello. Tale pratica cessò nel '58 e, conseguentemente, scomparve negli anni la
manifestazione della malattia.
• Sindrome di Gerstmann-Straussler-Scheinker (GSS)
Rara forma umana di TSE di tipo ereditario (autosomico dominante) ma trasmissibile
per inoculazione intracerebrale a cavie di laboratorio. È causata da una mutazione del
gene codificante la PrP ed è stata trovata in 50 gruppi famigliari. È caratterizzata da
demenza, atassia, disfagia, amiotrofia aggravantesi sino all'exitus, che sopraggiunge
dopo 2-6 anni dalla comparsa dei primi sintomi.
• Insonnia Familiare Fatale (FFI)
La FFI è una rara malattia ereditaria di tipo autosomico dominante come la sindrome
precedente ma caratterizzata da insonnia, disturbi del sistema nervoso autonomo,
disturbi motori e cognitivi e morte entro un anno dall’esordio.
• Malattia di Creutzfeldt-Jackob (CJD)
Può essere di tipo sporadico per via ignota (un caso su un milione), di tipo ereditario per
mutazione del gene codificante la proteina prionica o, in rari casi, di tipo iatrogeno: tali
casi compaiono in seguito ad infezione accidentale dovuta a procedure mediche
coinvolgenti materiale derivato da SNC infetto o ferri chirurgici non correttamente
decontaminati.
Inequivocabili caratteristiche cliniche sono la demenza rapidamente progressiva ed
almeno due dei seguenti segni clinici: mioclono, disturbi visivi o segni cerebellari,
piramidali o extrapiramidali, mutismo acinetico ed un tracciato elettroencefalografico
tipico. L'identificazione della proteina 14-3-3 nel liquido cefalorachidiano è di supporto
alla diagnosi.
• Variante della CJD (vCJD)
Questa forma si differenzia dalla forma classica di CJD per la comparsa anche in
soggetti particolarmente giovani, per una più lunga durata clinica della malattia
(superiore ad un anno) e per caratteristici sintomi d’esordio rappresentati da disturbi
comportamentali, modificazioni della personalità e depressione. La maggior parte dei
pazienti sviluppa precocemente un'atassia cerebellare, mentre, con il progredire della
malattia, può comparire mioclono preceduto da movimenti coreici; la demenza infine
evolve in un mutismo acinetico. Il quadro EEGrafico non presenta le caratteristiche
tipiche riscontrate nel CJD. All'esame neuropatologico si osservano numerosi depositi
di placche amiloidi circondate da spongiosi (placche floride). Ad oggi in Italia non è
stato segnalato alcun caso di vCJD.
46 _______________________________________________________________________________________
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Illustrazione schematica delle più frequenti localizzazioni delle lesioni da malattie prioniche. Nella BSE è colpito il midollo allungato, nella FFI la
regione talamica, nel CJD la corteccia cerebrale, mentre nel KURU e nella GSS viene maggiormente interessato il cervelletto.
ASPETTI ISTOPATOLOGICI COMUNI
1. Vacuolizzazione neuronale a livello dei processi dendritici, assonali e corpi cellulari
2. Ipertrofia e proliferazione astrogliale
3. Alterazione spongiforme della sostanza grigia
DIFFERENZIAZIONE DALLE PATOLOGIE INFETTIVE CONVENZIONALI
1. Assenza di reazione infiammatoria con assenza di manicotti perivascolari e
perineuronali, assenza di pleiocitosi o di un marcato incremento di proteine nel
liquido cerebrospinale durante il corso della malattia
2. Assenza di reazione immunitaria nei confronti dell'agente eziologico
3. Assenza al microscopio elettronico di particelle simil-virali nelle sezioni di encefali
colpiti
_______________________________________________________________________________________ 47
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Le ipotesi eziologiche
Sulla scorta di un recente lavoro di Bastian (Sito CJD Disease Foundation, 1999),
forniamo una rassegna delle ipotesi finora formulate, comprese quelle che hanno ormai perso
consistenza interpretativa (studi iniziali).
Anche se la teoria maggiormente accreditata è attualmente quella proposta da
Prusiner (1998) a cui si dà più ampio spazio, proponiamo anche una sintesi di altre teorie
recenti, che contengono comunque elementi di analisi interessanti.
Studi iniziali
• Teoria dei Virus Lenti (Sigurdsson, 1954; Gajdusek,1977)
Il termine virus lento fu proposto per denominare l'agente della scrapie basandosi sul
suo lungo periodo di incubazione: usare tale denominazione fu conveniente per la
caratteristica fisica che ai tempi poteva essere facilmente misurata, la dimensione, che in
seguito a prove di filtrazione fu determinata intorno ai 40nm. Contestualmente fu coniato il
termine di agenti non convenzionali per tener conto delle proprietà inconsuete degli agenti
delle TSE . Questa teoria fu abbandonata poichè nessuna particella che potesse essere
ricondotta all'aspetto di un virus convenzionale, è mai stata osservata al microscopio
elettronico; inoltre studi recenti non hanno mai evidenziato la presenza di acidi nucleici.
• Ipotesi del Viroide (Alper, 1967)
Il concetto di viroide scaturì dalla scoperta di proprietà attribuite all'agente infettante
che suggerivano una struttura più semplificata e più piccola dei virus convenzionali. A seguito
di studi di inattivazione con radiazioni, pareva che l’agente fosse una molecola di RNA di
200-400 nucleotidi priva di proteine (viroide). La teoria cadde quando si comprese che la
infettività era strettamente correlata alla presenza di proteine.
Teorie piu' recenti
• Teoria del Virino (Dickinson,1988)
L'ipotesi del virino è basata sul concetto che il patogeno sia una particella infettante
che possiede un genoma indipendente dal suo ospite; tuttavia, si ipotizzò che nel ciclo
replicativo della scrapie ci fosse uno stadio in cui il genoma fosse legato alle proteine
dell'ospite e necessitasse di un enzima (codificato da un gene dell'ospite) modulante i tempi di
replicazione. Questa teoria sebbene innovativa è essenzialmente speculativa, con poche o
nulle prove sperimentali a suo favore.
• Ipotesi del Virus Filamentoso (Merz, 1984)
Il ritrovamento di fibrille amiloidi associate a scrapie (SAF) nei tessuti colpiti da
CJD portò all'idea che queste fossero l'agente infettivo stesso, sulla base di una comparazione
tra le SAF presenti negli omogenati cerebrali e i batteriofagi filamentosi. Il primo problema
legato a questa ipotesi fu l'evidenza che il diametro delle fibrille (4-5 nm) risulta troppo
ridotto per contenere acidi nucleici: infatti la misura richiesta per la presenza di un genoma è
di almeno 20 nm. Inoltre in natura non vi sono altri esempi di strutture virali così peculiari.
Studi recenti hanno dimostrato che l'infettività non è assolutamente legata alle SAF.
48 _______________________________________________________________________________________
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
• Ipotesi del Retrovirus (Haseltine, 1986)
In prove di purificazione dell'agente eziologico della CJD, si evidenziò la presenza di
un RNA a basso peso molecolare simile a quello presente nei retrovirus. D'altra parte, la
provata assenza nei tessuti infetti da TSE di strutture virali evidenziabili al microscopio
elettronico, la resistenza ai trattamenti fisico-chimici che normalmente inattivano i retrovirus
e il diverso tipo di lesioni indotte nei tessuti da tali virus, permettono di scartare quest’ipotesi.
• Teoria della Amiloidosi-Virus-Indotta (Braig, 1985)
Poiché non esiste prova morfologica che un virus sia presente nei tessuti colpiti da
CJD, fu ipotizzato che siano visibili gli effetti di un virus piuttosto che il patogeno stesso. Le
SAF rappresenterebbero una struttura proteica amiloide connessa ad un'infezione virale,
ipotizzando che la replicazione del virus cominci e finisca prima dell'instaurarsi dei sintomi
clinici. Ciò giustificherebbe l'inefficacia del trattamento delle TSE con farmaci antivirali.
Questa teoria è stata indebolita dall'osservazione che le placche amiloidi sono state
scarsamente osservate nella scrapie indotta sperimentalmente, se non combinando l'agente
infettante di alcuni tipi di scrapie con certe linee di topi da laboratorio.
TEORIA MAGGIORMENTE ACCREDITATA
• Teoria del Prione (Prusiner, 1998)
Nel 1982 S.B. Prusiner pubblicò su Science uno studio sull'agente causale della
scrapie, affermando che fosse "una nuova particella proteica infettiva" e proponendo il nuovo
termine prione che teneva conto della sua natura sia proteica sia infettiva. L'idea di una
proteina come agente infettivo era stata già proposta da Griffiths nel 1967, ma fu Prusiner che
arrivò ad una vera e propria dimostrazione, tentando di capire perché, su 18.000 pecore
vaccinate contro il virus della louping-ill, circa 1.500 svilupparono dopo 2 anni la scrapie,
nonostante il vaccino fosse stato trattato con formalina per prevenire le infezioni virali. I suoi
esperimenti portarono all'evidenza che l'agente della scrapie conteneva una proteina
indispensabile all'infettività e le prove dimostrarono che l’agente perde la capacità infettante:
1. con la digestione mediante proteinasi K e tripsina;
2. per inattivazione chimica con dietil pirocarbonato;
3. per inattivazione con sodio dodecil solfato (SDS);
4. per inattivazione con sali come il guanidin tiocianato;
5. per inattivazione con fenolo;
6. per inattivazione con urea.
Inoltre l'agente si dimostrò resistente alle procedure d’attacco agli acidi nucleici
come il pH acido, l’azione di ribonucleasi, desossiribonucleasi, UV a 254 nm, l’idrolisi con
cationi bivalenti e l’attacco chimico con idrossilammina.
Si pensò che il peso molecolare dell'agente infettivo fosse tra 64.000 e 150.000
dalton, fatto incompatibile con la presenza di acidi nucleici o un sistema efficiente per la loro
riparazione; inoltre, recenti esperimenti di filtrazione ed elettroforesi portano a ritenere che
l'agente della scrapie abbia aspetto globulare ed abbia un peso molecolare addirittura inferiore
ai 50.000 dalton.
Le tipiche caratteristiche dell'agente della scrapie furono allora così riassunte:
1. stabile a 90° C per 30 minuti;
_______________________________________________________________________________________ 49
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
2. basso peso molecolare (inferiore ai 50.000 dalton);
3. contenente proteine idrofobe indispensabili all'infettività;
4. resistente alla ribonucleasi e desossiribonucleasi;
5. resistente alla radiazione UV a 254 nm;
6. resistente alla formazione di psoralene fotoindotta;
7. resistente all'idrolisi catalizzata da Zn bivalente;
8. resistente all'attacco chimico con NH2OH.
Fu così evidenziato che l'agente della scrapie differiva profondamente dai virus, i
viroidi e i plasmidi e, di conseguenza, fu proposto che avesse natura proteica e mancasse di
acido nucleico.
Contestualmente si cercò di provare che la proteina prionica, non solo fosse presente
in concomitanza con i sintomi della malattia, ma che ne fosse effettivamente la causa; già
dall'inizio degli studi sulla scrapie e sul kuru si era evidenziata la loro evidente trasmissibilità,
confermata dalla possibilità di infettare gli scimpanzè con inoculazione intracerebrale di
materiale infetto da CJD. La prova inoppugnabile fu data da Prusiner che dimostrò, tramite
prove di ultrafiltrazione, che il prione era l'unica macromolecola che potesse veicolare
l'infettività.
Dagli studi ipotizzanti la natura dei prioni è emerso quindi che l'agente infettivo era
composto da un singolo tipo di molecola proteica che fu chiamato PrPsc (scrapie) e,
successivamente, che tale proteina è la forma alterata di una normale glicoproteina di
membrana detta PrPc (cellulare).
Tabella comparativa tra Virus classici e Prioni
Virus Prioni
Agenti infettivi filtrabili Si Si
Presenza di acidi nucleici Si ??
Morfologia definibile con il Si No
microscopio elettronico
Presenza di proteine Si Si
Inattivazione con:
Formaldeide Si No
Proteasi Alcuni No
Calore (80°C) Quasi tutti No
Ionizzazione e raggi UV Si No
SINTOMI
Effetto citopatologico Si No
Periodo di incubazione Dipende dal virus Lunga
Risposta immunologica Si No
Produzione di interferone Si No
Risposta infiammatoria Si No
50 _______________________________________________________________________________________
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Caratteristiche della PrP
(Harris, 1999)
PrPc
CARATTERISTICHE E FUNZIONE (Quaglio, 2001)
La PrPc è una normale glicoproteina cellulare presente nei neuroni, nella glia del
SNC centrale, come pure in parecchi tessuti periferici, nei leucociti e nelle cellule spermatiche
mature (Shaked, 1999). La PrP e il suo RNAm sono ampiamente distribuiti nel sistema
nervoso centrale ed in particolare nei neuroni della corteccia, dell'ippocampo, nelle cellule
cerebellari del Purkinje e nei motoneuroni spinali. La normale funzione della PrPc rimane
sconosciuta, sebbene la sua localizzazione sulla superficie cellulare faccia pensare ad un suo
ruolo nell’adesione e nel riconoscimento cellulare, nei recettori di membrana e nella chimica
dei neurotrasmettitori. La comprensione del ruolo fisiologico della PrPc potrebbe essere
importante per capire la patogenesi della malattia, poiché la proteina può cessare di svolgere
la propria funzione quando si converte nell'isomero PrPsc. I topi knockout (animali in cui è
stato distrutto il gene codificante la PrPc) non dimostrano rilevanti difetti di sviluppo o di
comportamento: studi hanno evidenziato tuttavia, in alcuni fenotipi e con manifestazioni di
modesta gravità, sintomi che ricordano quelli delle malattie prioniche, come perdita del ritmo
circadiano, anomalie del sonno e cambiamenti dell’apprendimento. Recentemente è stato
proposto che la PrPc abbia un ruolo nel metabolismo del Cu poiché lega da uno a tre atomi
covalenti di rame a livello del N-terminale della regione ottapeptidica e mostra, infatti,
un’attività antiossidante: studi in vitro hanno dimostrato che la PrPc protegge le cellule dallo
stress ossidativo quanto più risulta legata ad atomi di rame (Brown e coll., 2001). Inoltre, in
omogenati cerebrali, tale proteina è stata rilevata in alta concentrazione nella giunzione
sinaptica postulando per questo una sua azione o come tampone del rame o come recettore
endocellulare per la reintroduzione del rame dall'ambiente extracellulare (Kretzschmar e coll.,
2000).
STRUTTURA E BIOSINTESI (Rymer e Good, 2000)
La PrPc è una proteina di circa 250 aminoacidi; presenta un’estremità
carbossiterminale che permette l'ancoraggio alla membrana legandosi al glicosil-
fosfatidilinositolo (GPI). Tra gli aminoacidi 178 e 213 la PrPc possiede due siti di
glicosilazione in prossimità di due residui di asparagina (N). Agli estremi di questo segmento
vi sono due residui di cisteina che formano un ponte disolfuro, legando tra di loro due tratti
della proteina che si dispongono nello spazio secondo una struttura elicoidale (alfa-elica).
Muovendosi verso l'estremità aminoterminale la proteina presenta un nucleo idrofobico
contenente un sito di scissione (clivaggio B) dove la proteina viene idrolizzata durante il
normale metabolismo cellulare. Sono di seguito presenti sequenze aminoacidiche ottameriche
ripetute, che possono legare fino a 4 atomi di rame; infine, vi è un’estremità N-terminale
chiamata "peptide segnale".
La struttura terziaria della proteina mostra una lunga (aa dal 23 al 121) e flessibile
coda N-terminale, tre alfa eliche (denominate elica 1, elica 2 ed elica 3) e due piccoli tratti
conformati a beta foglietto affiancati alla prima alfa elica.
Come molte proteine di membrana, la PrPc è sintetizzata nel reticolo endoplasmatico
ruvido (RER) e transita nell'apparato del Golgi per raggiungere la superficie cellulare.
_______________________________________________________________________________________ 51
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Fig. 2 : struttura della PrPc (sovrapposizione che mette a confronto quella del criceto e quella del topo)
Fig. 3 : Struttura della PrPc in soluzione ottenuta da Wüthrich, Glockshuber, and coll. al Swiss Federal Institute
of Technology . Si notano le tre alfa eliche, i foglietti beta presso il carbossiterminale della proteina,e il
segmento in "disordine flessibile" all'N-terminale.
52 _______________________________________________________________________________________
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Durante la sua biosintesi, la PrPc è soggetta a molte modificazioni come la scissione
del peptide segnale, l’unione di due siti N a catene oligosaccaridiche, la formazione di un
unico legame S-S e l’attacco al sito di ancoraggio del GPI.
Molti lavori condotti sul metabolismo della PrPc hanno dimostrato che essa viene
normalmente sottoposta a due scissioni o clivaggi. Uno avviene a livello dell'ancoraggio al
GPI, con conseguente rilascio della catena polipeptidica nell'ambiente extracellulare,
probabilmente tramite l'intervento di una fosfolipasi di membrana; il secondo è di tipo
proteolitico e si è osservato in una porzione di 16 aminoacidi della sezione idrofobica (sito di
clivaggio B). Entrambi i clivaggi hanno luogo tardivamente rispetto all'emivita della proteina
e sono state ritrovate proteine a diversi stadi di trasformazione oltre alla forma integra.
Nonostante gli studi compiuti, il significato fisiologico di queste scissioni è ancora
sconosciuto, forse, se la PrP serve da recettore di membrana, il clivaggio può rappresentare un
meccanismo di modulazione del recettore.
__ S – S __
I I II
II
II
II
II
C B A
Peptide segnale Segnale per il GPI
Ripetizioni ottameriche Ancoraggio al GPI
Zona idrofobica Oligosaccaridi legati a N
S–S Legame disulfidico
Fig, 4 - Struttura e processi post-translazionali della PrP: in alto la struttura primitiva della PrP dei mammiferi,
sotto forma matura della proteina. L'ancoraggio al GPI unisce il polipeptide alla membrana; le frecce A e B
indicano i siti di scissione nella PrPc e la freccia C indica il sito di scissione nella PrPsc. Il sito A risiede
all'interno dell'ancoraggio al GPI.
_______________________________________________________________________________________ 53
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
LOCALIZZAZIONE SUBCELLULARE
La PrPc è una proteina di superficie cellulare e, poiché è ancorata al GPI, può essere
rilasciata trattando la cellula con una fosfolipasi specifica (PI-PLC) a livello della porzione
diacilglicerolica.
La dimostrazione biochimica che la PrPc nei neuroni è trasportata assonalmente sino
alla porzione terminale dei nervi, è la presenza della proteina stessa sulle superfici delle
sinapsi (come osservato mediante prove di immunofluorescenza al microscopio elettronico).
Con l'immunoistochimica si è potuto evidenziare che la PrPc è principalmente concentrata
nelle sinapsi del bulbo olfattorio, limbo e sostanza nigro-striata, confermando la sua possibile
funzione all'interno della sinapsi. Diversi studi hanno dimostrato che, comunque, la PrPc non
permane sulla superficie cellulare ma piuttosto compie un ciclo tra la membrana cellulare ed il
citoplasma; si è osservato, in cellule neuronali in vitro, che il ciclo delle molecole di PrP si
compie in circa 60 min. e durante ogni passaggio dall'1 al 5% di tali molecole sono scisse al
sito B. Il sistema di riciclaggio della PrPc sembrerebbe interessante per due motivi. Da un
lato, potrebbe essere il momento in cui avviene la trasformazione della proteina normale in
PrPsc, dall’altro suggerirebbe che una delle funzioni fisiologiche della PrPc sarebbe quella di
recettore per l'introduzione di uno ione extracellulare, che recenti studi identificano con il
rame. Si è ipotizzato che la PrPc leghi il rame sulla superficie cellulare, lo trasporti in un
organo endocitico ove gli ioni, una volta dissociati da essa, vengono trasportati nel citosol da
altre proteine carrier del rame; la PrPc quindi ritornerebbe sulla superficie cellulare per
ricominciare il ciclo. Ciò sarebbe confermato dal fatto che una alta concentrazione di ioni
rame stimola rapidamente e reversibilmente l'endocitosi della PrP dalla superficie della cellula
(Pauly e Harris, 1998).
PrPsc
CARATTERISTICHE E LOCALIZZAZIONE SUBCELLULARE
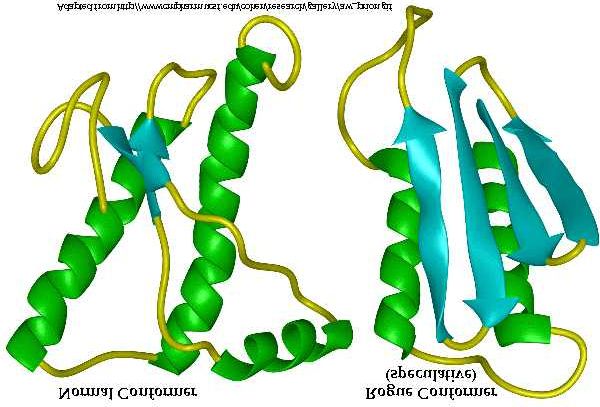
La conversione della PrPc nella forma PrPsc è essenzialmente di tipo
conformazionale, avendo le due isoforme identica sequenza aminoacidica. Il cambio di
conformazione comporta un sostanziale incremento della quantità di foglietti beta e una
leggera diminuzione delle alfa eliche: mediante prove spettroscopiche si è dimostrato che la
PrPc contiene il 42% di alfa eliche e una bassissima presenza di beta foglietti (3%); questi
ultimi divengono per la PrPsc il 43%, mentre il contenuto di alfa eliche è inferiore (30%)
(Pan, 1993). Sebbene la struttura terziaria della PrPsc sia stata solo ipotizzata, la teoria più
attuale sulla produzione di questo isomero comporterebbe in primo luogo la modifica dell'N
terminale posto a metà della proteina, ma anche la trasformazione di parte della coda N-
terminale in foglietto beta, essenzialmente tra i residui 90-121 e forse, in parte, dell'alfa-elica
1.
La localizzazione subcellulare della PrPsc non è stata attualmente ben determinata a
causa della sua scarsa immunoreattività; l’inconveniente potrebbe essere ovviato con il
trattamento mediante agenti denaturanti che però agiscono sulla morfologia cellulare,
alterandola. Pare che la PrPsc sia presente sia nel Golgi sia ancorata al GPI della membrana
cellulare, analogamente alla PrPc, tramite un C-terminale. Tale legame però non è sensibile
all'azione della fosfolipasi specifica PI-PLC, suggerendo che l'ancoraggio sia di tipo diverso
rispetto alla PrPc. E' stato ipotizzato che lo stesso cambiamento strutturale che rende la PrP
alterata parzialmente inattaccabile alla proteasi K, possa renderla resistente alla PI-PLC.
54 _______________________________________________________________________________________
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Mediante prove sperimentali su cellule geneticamente modificate (CHO) che
producono una PrP mutante, è stato possibile stabilire che il processo di conversione che porta
alla PrPsc avviene in tre stadi successivi: il primo e quasi immediato cambiamento
biochimico porta all'acquisizione della resistenza alla PI-PLC, il secondo, osservabile dopo
almeno 1 ora dall'inizio del processo, comporta l'insolubilità ai detergenti dovuta
presumibilmente all'aggregazione delle molecole. Il terzo stadio, evidenziabile sino a 7 ore
dopo, rende la molecola resistente alle proteasi, per ulteriori processi di aggregazione e
polimerizzazione.
Da esperimenti compiuti su colture cellulari si è riscontrato che, una volta
sintetizzata, la PrPsc risulta metabolicamente stabile per periodi variabili da 24 a 48 ore e
tende ad un lento accumulo, in contrasto con la PrPc il cui ciclo ha un'emivita tra le 4 e le 6
ore. Comunque, solo una minima quantità di molecole di PrPc viene trasformata in PrPsc. Le
rimanenti subiscono un metabolismo esattamente sovrapponibile a quello delle cellule non
infette: anche se le vie metaboliche della PrPc non sono ancora state del tutto chiarite, pare
che vi siano coinvolti gli endosomi ed i lisosomi.
Fig. 5: sulla sinistra modello della PrP normale (cellulare), sulla destra ipotetica conformazione della PrPsc
anomala (rogue)
Tabella di confronto tra PrPsc e PrPc
PrPsc PrPc
Struttura Globulare Estesa
resistenza alle proteasi Si No
Presenza nelle fibrille scrapie Si No
associate
Turnover giorni Ore
_______________________________________________________________________________________ 55
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
MODIFICHE STRUTTURALI E INTERAZIONI FISICHE
Si pensa che durante l'infezione prionica avvenga una interazione fisica altamente
specifica tra la PrPc e la PrPsc responsabile della produzione di nuove molecole anomale.
Questa conclusione si basa su parecchie prove. In particolare, i topi knockout sono
completamente resistenti all'infezione prionica, mentre topi transgenici, esprimenti PrP
normale di criceto, sono suscettibili all'infezione derivata dal criceto, cosa che non avviene
nei topi non modificati geneticamente. Altri studi hanno provato che alcuni segmenti della
PrPc sono indispensabili per la trasformazione in PrPsc. Infatti, il grado di omologia
aminoacidica della regione centrale della proteina influenza profondamente l'efficienza del
processo di trasformazione; staccando un singolo residuo aminoacidico da questa regione si
può prevenire la formazione di PrP alterata. Tale squisita specificità fa pensare che esista una
forte barriera contro la trasmissione interspecifica delle malattie da prione.
È stata dimostrata in vitro la trasformazione della PrPc in PrPsc ma la quantità
iniziale di PrPsc necessaria per innescare la reazione era eccessiva rispetto a ciò che accade in
vivo. Inoltre, la PrPsc così ottenuta non aveva potere infettante (Kocisko e coll., 1995).
Questo fa pensare che nella cellula sia presente un fattore assente nei sistemi sperimentali
purificati e che ciò sia la causa della scarsa efficienza stechiometrica della reazione in vitro.
IPOTESI SULLA FORMAZIONE DELLA PrPsc
Vi sono attualmente due teorie sulla trasformazione di PrPc in PrPsc.
La prima detta "nucleated polymerization" ipotizza che la polimerizzazione della
molecola inizi da un nucleo di PrPsc preesistente a cui si legano monomeri di PrPc: cioè la
PrP anomala potrebbe "catalizzare" la formazione di un polimero alterato. Questo processo
pare simile a quelli che avvengono in natura per altri tipi di proteine quali: la
polimerizzazione della tubulina, la crescita dei cristalli, la formazione di Hb falciforme,
l'assemblaggio del capside virale e soprattutto la polimerizzazione dei flagelli dei batteri.
Fig. 6 - Schema della teoria del "template assistance"
56 _______________________________________________________________________________________
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
La seconda teoria detta a stampo o "template assistance" postula che un numero
relativamente piccolo di molecole di PrPc e di PrPsc formino un oligomero tramite un
ipotetico chaperon molecolare. I componenti della PrPsc funzionano come uno stampo che
imprime la sua conformazione alla PrPc o ad un suo intermedio, parzialmente modificato
chiamato PrP*. In questo caso la barriera cinetica tra i due isomeri è superata dall’azione
catalitica dell'ipotetico composto PrPsc più chaperon.
Anche per quest’ipotesi esistono osservazioni fatte su altre proteine esistenti in
natura quali: l'emoagglutinina del virus influenzale, inibitori delle proteasi (serpine), ed un
certo numero di proteasi, quali la subtilisina e le proteasi alfa-litiche.
Questi due modelli comunque, non si escludono a vicenda perché potrebbe esserci un
meccanismo ibrido nel quale la superficie di un aggregato, inizialmente formatosi per un
processo di nucleazione, funge da stampo su monomeri di PrPc. Tali teorie, inoltre, sono
applicabili sia alle forme genetiche come pure a quelle infettive, poiché la presenza di una
mutazione patologica presumibilmente favorirebbe la formazione spontanea o di nuclei di
PrPsc o di PrP*.
RUOLO DEGLI CHAPERON MOLECOLARI
Gli chaperon (letteralmente: chi introduce qualcuno in un ambiente nuovo) sono proteine
che facilitano il ripiegamento dei polipeptidi durante la loro biosintesi ed il loro
trasporto negli organuli intracellulari, aiutando a prevenire l'aggregazione delle
proteine durante condizioni di stress cellulare come lo shock da calore. Si pensa che essi
agiscano legandosi ai loro substrati, con un’azione talvolta ATP dipendente, prevenendo
la formazione di forme intermedie inattive. Nella cellula gli chaperon sono stati ritrovati
in quasi tutti i settori cellulari; la PrP, durante il suo ciclo di formazione, passa solo nel
RER dove, per altro, sono state sicuramente individuate molecole-chaperon. Poiché la
formazione di PrPsc comporta una modifica del ripiegamento delle proteine e la
formazione di aggregati, processi nei quali intervengono gli chaperon, è stata formulata
l'ipotesi che essi vi svolgano un ruolo. Sono stati compiuti un certo numero di
esperimenti per supportare il ruolo degli chaperon nella biologia dei prioni. Si è notato
che topi geneticamente modificati in modo da esprimere la PrP del criceto sviluppavano
la malattia prionica dei criceti cosa che non avviene nei topi con il gene della PrP
umana (Hu PrP) che si dimostravano insensibili ai prioni umani. Tuttavia, topi chimerici
geneticamente esprimenti una proteina prionica mista topo/uomo (Hu/Mo PrP) erano
sensibili ai prioni umani come pure gli incroci tra topi HuPrP e topi knockout. Questi
risultati sono stati interpretati ipotizzando l'esistenza di chaperon cellulari chiamati
genericamente proteina X, che interagirebbero in modo specie-specifico con il segmento
C-terminale della PrPc. Questa potrebbe anche essere la spiegazione della propagazione
elettiva dei prioni in particolari popolazioni neuronali e in alcuni tipi di cellule
periferiche come quelle del sistema linforeticolare. Ci sono prove che chaperon
intervengano nella biogenesi dei prioni in colture in vitro di cellule di neuroblastoma
infettate da scrapie. D’altro canto parecchi chaperon "chimici", come il glicerolo e il
dimetilsulfossido, che stabilizzano la conformazione delle proteine, inibiscono la
produzione di PrPsc in cellule infettate, le quali mostrano anche una diminuita resistenza
allo shock da calore. La prova più diretta che gli chaperon possono accelerare la
produzione di PrP anomala, è stata fornita da esperimenti nei quali due di queste
molecole (una derivata da lieviti Hsp104 e l'altra di origine batterica GroEL) hanno
aumentato la produzione di PrPsc in un substrato in cui non erano presenti elementi
cellulari (DeBurman, 1997).
_______________________________________________________________________________________ 57
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Meccanismi patogenetici delle TSE
Mancano pressoché totalmente dati patogenetici riguardanti la BSE. Le uniche
informazioni disponibili sono relative alla distribuzione dell’infettività nei tessuti, esito dei
lavori di infezione sperimentale condotti da autori inglesi non soltanto sui topi, ma anche su
animali della specie bovina.
Per il resto la maggior parte delle conoscenze è desunta dalle ricerche condotte sui
roditori, e precisamente sui topini infettati sperimentalmente con l’agente della scrapie, o da
studi svolti su pazienti affetti da vCJD.
Tutte le ricerche hanno comunque dimostrato che, a seguito di un’infezione
attraverso vie indirette (via orale in particolare), si verifica un lungo periodo di eclissi, durante
il quale non è rilevabile infettività in alcun tessuto. Ciò lascia presumere l’esistenza di
meccanismi e sedi di replicazione non ancora ben individuati. I tessuti linforeticolari quali
tonsille, milza, linfonodi e, in caso di contagio per via orale, il tessuto linfoide annesso
all’intestino (placche di Peyer) sembrano essere la prima sede di replicazione. Un altro
possibile serbatoio potrebbe essere rappresentato da alcuni distretti del SNP e precisamente da
alcuni gangli sensitivi e autonomi (gangli enterici e gangli delle radici dorsali) (Mc Bride e
coll., 1999; Groschup e coll., 1999; Glatzel e Aguzzi, 2000). Per quanto concerne la BSE va
sottolineato che sembra di scarso rilievo la presenza dell’agente infettante a livello splenico,
come sede di sviluppo e replicazione (Aguzzi, 2001). A tal riguardo risulterebbero più
importanti i tessuti linfo-reticolari come le amigdale e le placche di Peyer annesse
all’intestino.
La neuroinvasione è una fase critica che è stata oggetto di numerosi studi da parte di
diversi gruppi di ricercatori. Kimberlin e coll. (1983) hanno studiato in particolare l’infettività
dei diversi tessuti in topini sperimentalmente infettati da scrapie. Diringer e coll. (cit. in Czub
e coll, 1986), focalizzando la loro attenzione sul ruolo del sistema nervoso periferico nella
neuroinvasione, hanno valutato l’infettività negli hamster. Più recentemente alcuni ricercatori
del gruppo di Aguzzi hanno concentrato la loro attenzione sulla patogenesi periferica
attraverso lo studio delle modalità di neuroinvasione dei prioni nei topini.
Tutti i ricercatori hanno stabilito il ruolo preponderante dei tessuti linforeticolari ed
in particolare delle cellule dendritiche follicolari (FDC) presenti nella milza, come nei
linfonodi e nelle placche di Peyer dell’intestino. Come dimostrato da numerose ricerche la
replicazione o almeno l’accumulo dei prioni in tali cellule dipende comunque particolarmente
dalla loro espressione della PrP cellulare (Bruce, 2001).
CELLULE FOLLICOLARI DENDRITICHE (FDC)
Sono presenti nella parte scura periferica del centro germinativo dei noduli linfatici ed
in altre sedi timo-dipendenti degli organi linfatici, come ad es. la zona marginale della
polpa bianca della milza ed i cordoni midollari dei linfonodi. Sono cellule grandi,
irregolarmente stellate, con lunghi prolungamenti ramificati connessi con quelli delle
cellule dendritiche vicine e con molti linfociti B.
Le FDC hanno la funzione di presentare gli antigeni ai linfociti: una volta incontrato
l’antigene, le FDC lo rielaborano, lo concentrano e lo espongono sulla superficie,
presentandolo ai linfociti. Svolgono quindi un importante ruolo immunostimolante, in
quanto favoriscono il contatto dei linfociti B del centro germinativo dei noduli linfatici
con gli antigeni stessi.
58 _______________________________________________________________________________________
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Anche i linfociti B sembrano avere un ruolo cruciale, ma per ora non del tutto chiaro.
Sembra che loro importanza sia essenzialmente legata al fatto che permettono la maturazione
e pertanto il raggiungimento della completa funzionalità delle FDC elaborando fattori di
maturazione quali le linfotossine alfa/beta di membrana (Montrasio e coll., 2000). Infatti,
studi svolti su topini hanno dimostrato che trattamenti con recettori solubili delle linfotossine
beta portano alla scomparsa delle FDC mature dalla milza. Tale trattamento impedisce anche
l’accumulo dei prioni nella milza, ritardando pertanto la neuroinvasione dopo inoculo
intraperitoneale di materiale scrapie-infetto (Montrasio e coll., 2000; Weissmann, 2001). Le
FDC sembrano pertanto il candidato più probabile per la replicazione dei prioni, perlomeno
nella milza. In tale distretto, infatti, dopo infezione sperimentale, forme abnormi di PrP si
accumulano tra i prolungamenti delle FDC, esattamente nei siti di ritenzione protratta degli
immunocomplessi. Da tali distretti presumibilmente la PrPsc è trasferita ai linfociti splenici
posti in intimo contatto con le FDC stesse. Relativamente al ruolo dei linfociti nel trasporto
dell’infettività, merita ancora di essere sottolineato il fatto che, se tali cellule (B e T) sono
infettanti a livello di milza, non sono invece stati sinora riscontrati linfociti infetti nel sangue
circolante (Raeber e coll., 1999; Montrasio e coll., 2000).
Nell’intestino pare poi che il superamento dell’epitelio da parte della PrP infettante
sia favorito da cellule epiteliali specializzate (M cells) il cui ruolo sembra sia il trasporto di
macromolecole e/o antigeni dal lume intestinale alla sottomucosa ovvero ai linfociti (Aguzzi,
2001).
CELLULE “M” O CELLULE EPITELIALI ASSOCIATE AI FOLLICOLI
Sono presenti qua e là nell’epitelio che ricopre le placche di Peyer (follicoli linfatici
aggregati nella sottomucosa dell’ileo.) Hanno pochi, irregolari, tozzi e lunghi microvilli
assai più grandi di quelli dell’orletto striato degli enterociti vicini. La membrana
plasmatica della faccia apicale presenta numerose e piccole introflessioni. Le facce
laterali e quella basale sono profondamente introflesse e contengono linfociti
intraepiteliali che hanno attraversato la membrana basale. Per tale ragione le cellule
“M” assumono l’aspetto di lamine sottili che circondano un piccolo gruppo di linfociti,
separandoli dal lume intestinale.
Si ritiene che le cellule “M” trasportino le macromolecole e/o gli antigeni
dal lume intestinale ai linfociti intraepiteliali o a quelli del tessuto linfatico sottostante,
in prevalenza di tipo B. Dopo aver ricevuto l’informazione antigenica i linfociti
intraepiteliali raggiungono i noduli linfatici e passano in circolo; ritornano quindi nei
noduli linfatici ed invadono la lamina propria della mucosa ileale: qui i linfociti B si
trasformano in plasmacellule che elaborano immunoglobuline A; queste attraversano le
cellule epiteliali rivestendosi di uno strato glicoproteico che ne evita la lisi da parte
degli enzimi proteolitici. Le immunoglobuline vengono quindi emesse alla superficie
dell’epitelio ileale proteggendolo dagli attacchi batterici.
Relativamente alle modalità di neuroinvasione sembra che diverse vie siano possibili.
La più probabile è una migrazione tramite i nervi periferici, soprattutto se la loro
mielinizzazione è ridotta o assente (Kimberlin e coll., 1983). Considerando che la zona
mantellare dei follicoli linfoidi è innervata da fibre non mielinizzate, questa modalità di
migrazione sembra ben adattarsi al modello. L’esatta modalità di trasporto non è nota:
sembrano possibili sia il trasporto assonale sia quello extra-assonale (Glatzel e Aguzzi, 2000).
_______________________________________________________________________________________ 59
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001CAPITOLO 2 ___________________________________________ EZIOLOGIA, PATOGENESI E DIAGNOSI
Circa i nervi coinvolti nella neuroinvasione le ipotesi sono diverse. Nel topo l’agente
infettante raggiunge il midollo spinale attraverso il sistema nervoso autonomo (nervo
splancnico). Dalla regione medio-toracica del midollo l’infezione diffonde poi caudalmente e
rostralmente (Kimberlin e coll., 1980; Cole e Kimberlin, 1985). Un’altra via possibile è il
nervo vago che sembra essere l’unica importante nell’hamster infettato oralmente (Beekes e
coll., 1996; Beekes e coll., 1998). Pertanto dagli studi sinora condotti pare possano essere
coinvolte sia fibre del simpatico sia fibre parasimpatiche (nervo vago).
Un ruolo importante nel trasporto dell’infettività, ma ancora da definire, riguarda
l’espressione della PrP cellulare (cfr. paragrafo 3).
E’ certo che l’agente infettante, dopo un lungo periodo di incubazione, raggiunge il
sistema nervoso centrale ove provoca le caratteristiche e ben note lesioni spongiformi
degenerative, la perdita di cellule neuronali e la gliosi.
Sui meccanismi mediante i quali l’agente infettante provoca le lesioni nervose sono
state formulate diverse ipotesi tra cui tre sono le principali:
a) Tossicosi da accumulo di PrP patologica (Hope, 2000; Hedge e coll., 1999);
b) Deplezione della PrP cellulare;
c) Attivazione della microglia.
a) Gli effetti neurotossici della PrPsc sembrano associati in vitro a particolari peptidi ben
definiti corrispondenti agli aminoacidi 105-132 e 106-126 rispettivamente della sequenza
murina e umana della PrP. La tossicità è stata dimostrata in colture di cellule neuronali e
in colture nervose miste, ove provocherebbe morte dei neuroni per apoptosi (Forloni e
coll., 1993), ipertrofia dell’astroglia e sua proliferazione (Forloni e coll., 1994; Hope e
coll, 1995). Alcuni lavori indicherebbero che il peptide non agisce direttamente sugli
astrociti, ma induce proliferazione della microglia, che a sua volta stimolerebbe la
proliferazione astrocitaria (Kretzschmar, 1995; Brown e coll., 1995).
Relativamente all’apoptosi sono stati condotti numerosi studi in vitro e in vivo con la
microscopia elettronica e l’ibridizzazione in situ, in particolare su topi sperimentalmente
infettati con scrapie (Kretzschmar, 1995; Giese e coll., 1995). Sembra particolarmente
accentuata a livello di cellule retiniche, strato dei granuli cerebellari, gangli basali, bulbo
olfattorio e corteccia cerebrale. Il fenomeno dell’apoptosi, che giustificherebbe peraltro la
sola reazione gliale e l’assenza di reazione infiammatoria, è comunque possibile solo se le
cellule esprimono la PrP cellulare (Brown e coll., 1995). Inoltre è strettamente associato
alla presenza di microglia in coltura: distruggendo la microglia la tossicità del peptide si
riduce notevolmente. Il peptide agirebbe pertanto sulla microglia, alterandone il
metabolismo e inducendo morte neuronale, probabilmente tramite attivazione di specifici
canali di trasporto del calcio (Brown e coll., 1995).
I fenomeni apoptotici neuronali sono stati più recentemente valutati anche nei bovini
affetti da BSE ove però non sono risultati molto evidenti; occasionalmente sono stati visti
invece nelle cellule gliali. L’apoptosi pertanto, perlomeno nella BSE, sembra non essere
uno dei meccanismi primari di perdita neuronale (Theil e coll., 1999).
b) Il ruolo della PrP cellulare sembra quindi in primo luogo indispensabile per consentire la
realizzazione dell’effetto tossico del peptide 106-126. Peraltro prove sperimentali
condotte sui topini deleti del gene codificante la PrP hanno dimostrato che la mancanza
della PrPc nei tessuti ostacola l’insorgenza di lesioni patologiche, a seguito di infezione
60 _______________________________________________________________________________________
MEDICINA VETERINARIA PREVENTIVA – Speciale 2001Puoi anche leggere

















































