CORRELAZIONI E SUSCETTIBILITA' ALLE MALATTIE DEGLI ALLELI DEL SISTEMA HLA - Core
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Università degli Studi di Pisa
Facoltà di Medicina e Chirurgia
Scuola di Specializzazione in Patologia Clinica
TESI DI SPECIALIZZAZIONE
CORRELAZIONI E SUSCETTIBILITA’
ALLE MALATTIE
DEGLI ALLELI DEL SISTEMA HLA
Candidata: Relatori:
Francesca Fantolini Prof. Aldo Paolicchi
Dottor Piero Palla
Anno Accademico
2013-2014
I
INDICE
RIASSUNTO pag 1
1. INTRODUZIONE pag 3
1.1 Sistema HLA pag 3
1.1.1. Antigeni HLA di classe I pag 3
1.1.2. Antigeni HLA di classe II pag 5
1.1.3. Nomenclatura Sistema HLA pag 8
1.2 Correlazione tra alleli HLA e malattie pag 11
1.2.1. Principali malattie autoimmuni associate al sistema HLA pag 16
1.2.2. Principali malattie infettive associate al sistema HLA pag 31
1.3 Scopo della tesi pag 34
2.MATERIALI E METODI
2.1 Microlinfocitotossicità complemento dipendente pag 36
2.1.1. Principio del test pag 36
2.1.2. Esecuzione test pag 38
2.1.3. Interpretazione risultati pag 40
2.2 PCR-SSP (sequence specific primer) pag 41
2.2.1. Principio del test pag 41
2.2.2 Esecuzione test pag 42
2.3 Metodiche alternative di tipizzazione HLA pag 45
3. INTERPRETAZIONE DEI RISULTATI pag 48
3.1 Microlinfocitotossicità complemento dipendente
per “spondilite anchilosante” e per “malattia Behçet” pag 49
3.2 PCR-SSP per “malattia celiaca”, per “diabete tipo I”,
per “pazienti HIV positivi” pag 51
4. DISCUSSIONE pag 57
RIFERIMENTI BIBLIOGRAFICI pag 59
II
RIASSUNTO
Negli ultimi anni sono state individuate numerose associazioni fra antigeni HLA e
malattie soprattutto a patogenesi immunitaria. Queste scoperte hanno contribuito a
delucidare i meccanici patogenetici di alcune patologie e a valutare l’utilità clinica
della tipizzazione HLA. In particolare sono state fornite indicazioni chiare su quali
loci e a quale risoluzione è corretto tipizzare tali malattie, in modo da favorire l’uso
delle risorse dei laboratori nel modo più razionale possibile.
In questo lavoro di tesi, svolto presso il laboratorio di tipizzazione tissutale HLA
dell’AUSL 6 di Livorno, sono stati presi in esame, nell’anno 2013, i pazienti
richiedenti la suscettibilità genetica per patologie associate al sistema HLA, in
particolare per la spondilite anchilosante, per la malattia celiaca, per il diabete
mellito di tipo I, per la malattia di Behçet e per valutare nei pazienti hiv positivi
l’eventuale insorgenza della reazione di ipersensibilità al trattamento con abacavir.
E’ stato impiegato il test di microlinfocitotossicità complemento dipendente (test
sierologico) per valutare l’eventuale presenza dell’aplotipo HLA-B27 per la
spondilite anchilosante e dell’aplotipo HLA-B51 per la malattia di Behçet, e la PCR-
SSP (test di biologia molecolare) per valutare l’espressione dell’eterodimero DQ2
e/o DQ8 per la celiachia, dell’aplotipo DR3 e DR4 per il diabete mellito di tipo I e
dell’allele HLA-B*57:01 per valutare la risposta al trattamento terapeutico con
abacavir nei pazienti HIV positivi.
Questi non possono essere considerati test né di routine o screening, ma possono solo
fornire un supporto diagnostico al clinico. L’unico fenotipo potenzialmente utile
nella diagnosi risulta la spondilite anchilosante, che presenta un’alta frequenza nei
pazienti affetti e una relativamente bassa nella popolazione normale; ha valore, ad
1
esempio nei casi ad esordio precoce di artrite con utilità per la classificazione delle
forme giovanili.
Per le altre patologie questo test ci consente di valutare solo la predisposizione
genetica alla malattia, ad esempio, per la celiachia, a seconda dell’allele espresso,
identificato con PCR in alta risoluzione, si identifica il “gruppo di rischio”, che
consente per i familiari di primo grado dell’affetto, sulla base del rischio, di definire
la frequenza degli esami sierologici da effettuare nel tempo, onde seguire con
massima attenzione la possibile insorgenza della malattia.
2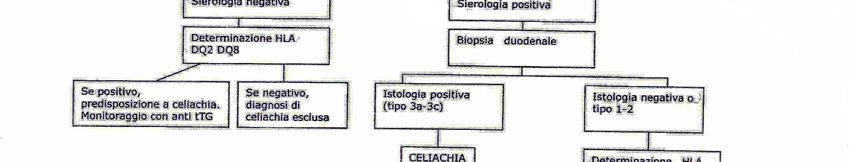
INTRODUZIONE
1.1 SISTEMAHLA
I geni del sistema HLA sono localizzati sul braccio corto del cromosoma 6 (6p21.1-
21.3, a 6000 Kb circa dal centromero) e rappresentano una parte della regione
genetica conosciuta come MHC, la cui funzione specifica è la regolazione della
risposta immune.
Questi geni si dividono in tre classi secondo il tipo di proteina che codificano, ma
solo sei loci, rispettivamente i loci HLA A, B, C (che codificano proteine di classe I)
e i loci DR, DQ e DP (che codificano proteine di classe II) codificano proteine che
costituiscono gli alloantigeni di istocompatibilità convolti nella presentazione
dell’antigene e quindi nel controllo della risposta immune. Tra il locus B ed il locus
DR si estende per 98 Kb la regione di classe III, che codifica per proteine solubili
come ad esempio le frazioni C2 e C4 del complemento.
FIG. 1 Struttura genetica dell’HLA
3
Il Complesso Maggiore di Istocompatibilità (MHC) ha sviluppato nel corso della sua
evoluzione tre caratteristiche fondamentali: polimorfismo, poligenia e codominanza.
E’ considerato un sistema polimorfico in quanto esistono numerosi alleli per ogni
gene e questo ha lo scopo di aumentare la variabilità nella composizione chimica
della “tasca”, di modo che nessuna molecola estranea possa sfuggire al meccanismo
di riconoscimento da parte dei linfociti T.
Per questo motivo ogni molecola di classe I e II è in grado legare peptidi diversi.
Infatti il legame peptide-molecola HLA non è specifico, ma è eterogeneo (ad una
molecola HLA possono associarsi più peptidi).
Infatti il polimorfismo a livello delle molecole di tipo I e II determina le varie
differenze immunogenetiche tra individui appartenenti alla stessa specie e
rappresenta il meccanismo che è alla base del fenomeno del rigetto dei trapianti.
E’ un sistema poligenico, ovvero vi sono numerosi geni che codificano per le
proteine di I e II classe, con diversa specificità per i vari peptidi.
Infine l’espressione degli alleli HLA è di tipo codominante: in ogni essere umano
risultano essere espressi i prodotti di entrambi gli alleli (uno di origine materna e uno
di origine paterna), quindi entrambe le molecole partecipano al meccanismo di
presentazione degli antigeni.
4
1.1.1 Antigeni HLA di classe I
Gli antigeni di classe I (HLA A, B, C) sono presenti sulla membrana di tutte le
cellule nucleate dell’organismo e sulle piastrine. Sono anche presenti, allo stato
solubile, nel sangue.
La molecola è costituita da un dimero proteico formato da una catena pesante
(catena α, PM 44 KDa) e da una catena leggera denominata β2-microglobulina (PM
12 KDa), unite tra loro da legami non covalenti. Delle due, solo la catena α, nella
quale risiede l’allotipia, è codificata da geni HLA. La β2-microglobulina è invece
controllata da geni posti sul cromosoma 15.
Lo studio chimico di questi antigeni ha permesso di ottenere informazioni precise
sulla loro struttura, in particolare è stato evidenziato che la catena α è costituita da tre
porzioni distinte: la prima, intracitoplasmatica, di 32 aminoacidi implicata nella
trasduzione del segnale; la seconda, sempre di 32 aminoacidi, transmembranaria
idrofobica probabilmente in una configurazione ad α elica; la terza
extramembranosa, di 247 aminoacidi. In quest’ultima porzione periferica sono inoltre
riconoscibili tre diversi “domini” (regioni) di uguale lunghezza denominati α1 (N-
terminale), α2 e α3. Il dominio α1, codificato dall’esone 2, è composto dai residui
aminoacidici 1-90; mentre i domini α2 (codificato dall’esone 3, residui aminoacidici
91-182) e α3 (codificato dall’esone 4, residui aminoacidici 183-274) hanno ponti
disolfuro intracatenari che comprendono regioni di 63 e 86 aminoacidi
rispettivamente e danno una conformazione ad anello. Il dominio α3 ha estese
omologie con la regione costante delle immunoglobuline, appartiene infatti alla
superfamiglia delle Ig.
5
I domini polimorfici α1 ed α2 interagiscono per formare una piattaforma costituita da
un foglietto β planare ad 8 strisce antiparallele, delimitato da due regioni ad α elica.
Quattro strisce del foglietto β ed una delle due α eliche sono costituite dai residui
aminoacidici del dominio α1, mentre le restanti sono costituite dai residui
aminoacidici del dominio α2. Ne risulta che i due domini esterni α1 ed α2 delimitano
un solco che prende il nome di “tasca combinatoria” , alla quale si lega il peptide che
presenta una lunghezza di 8-11 aminoacidi.
La parte più esterna della tasca è quella responsabile del contatto con il recettore dei
linfociti T, mentre la parte più interna è quella in cui si formano i legami con il
peptide.
La variabilità dei domini α1 e α 2 è fondamentale per permettere l’alloggiamento di
un alto numero di peptidi diversi e per l’interazione con il recettore dei linfociti T.
La regione α 3 invece non è polimorfica cioè non varia tra le molecole HLA-I e
contiene un’ansa che serve come sito di legame per i CD8 (linfociti T citotossici).
1.1.2 Antigeni HLA di classe II
Gli antigeni HLA di classe II (HLA-DR, HLA-DQ, HLA-DP) non hanno una
distribuzione ubiquitaria, ma sono presenti in quantità dimostrabile solo su alcuni tipi
cellulari: linfociti B, macrofagi, cellule di Langherans, alcune cellule endoteliali e
compaiono su altri tipi cellulari in condizioni di attivazione (per es. sui linfociti T).
Sono inoltre presenti allo stato solubile nel sangue circolante.
Nella sottoregione DR sono stati localizzati un gene, che codifica per la catena α
(gene A), che non è polimorfica, e quattro geni che codificano per la catena β
altamente polimorfica (geni B).
6
Le sottoregioni DP e DQ contengono entrambe un gene A e un gene B, che
codificano rispettivamente una catena α e una catena β, entrambe polimorfiche. In
ciascuna delle due regioni sono presenti un gene α e un gene β che non sono espressi
a livello fenotipico.
Da un punto di vista molecolare, questi antigeni sono dimeri costituiti da due catene
polipeptidiche α (PM 34 kDa) e β (PM 29 kDa) associate da legami non covalenti.
Entrambe le catene sono suddivise in tre regioni: -regione extracellulare idrofilica
composta da due domini α1,α2 per una catena e β1 e β2 per l’altra catena, -regione
idrofobica transmembrana con struttura ad alfa-elica e -regione idrofilica
intracitoplasmatica
La regione che lega il peptide è formata dall’interazione dei domini α1 e β1 delle due
catene, che formano una tasca con il pavimento e le pareti rispettivamente formate da
un foglietto β planare a 8 strisce antiparallele, delimitato da due regioni ad α elica.
Quattro strisce del foglietto β ed una delle due α eliche sono costituite dai residui
aminoacidici del dominio α1, mentre le restanti sono costituite da residui
aminoacidici del dominio β1. I peptidi, che si legano alla molecola HLA presentano
una lunghezza di circa 30 aminoacidi. Nell’uomo il polimorfismo maggiore si ha per
il dominio β1, mentre i domini α2 e β2 non sono polimorfici.
La regione β2 costituisce il sito di legame dei CD4 (linfociti Thelper).
7
Fig.2 Rapprentazione molecole HLA di I classe e di II classe
1.1.3 NOMENCLATURA SISTEMA HLA
I geni MHC sono i geni più polimorfi presenti nel genoma umano e in quello di tutte
le specie finora analizzate. Un’analisi svolta con approccio sierologico eseguita
sull’uomo ha portato all’identificazione di più di 150 alleli HLA diversi, ma con
l’uso di nuove tecniche più sensibili e specifiche si è visto che ogni allele può avere
numerose varianti non rilevabili con il test sierologico.
Tab. 1 Alleli HLA classe I secondo l’IMGT/HLA Database dell’Antony Nolan Istitute
8Tab. 2 Alleli HLA classe II secondo l’IMGT/HLA Database dell’Antony Nolan Istitute
mutazione allele null
fuori reg. codificante
prefisso del gene gene gruppo di specifica mutazione mutazione
alleli proteina sinonima fuori reg.cod
Fig.3 Nomenclatura HLA
Ciascun allele HLA è definito da un “codice” che ne indica la specificità. Questo
codice è costituito da coppie di numeri definite “digits”, le tipizzazioni possono
essere definite a 2, 4 o 6 digits.
I primi due digits indicano vari tipi e nella maggior parte dei casi corrispondono agli
antigeni sierologici caratterizzanti un allotipo.
9Il terzo ed il quarto digits caratterizzano i sottotipi antigenici e sono assegnati sulla
base della loro identificazione genetica mediante sequenziamento diretto degli
antigeni.
Gli alleli che si differenziano nei primi 4 digits differiscono in uno o più posizioni
nucleotidiche e quindi nella sequenza amminoacidica della proteina codificata.
Il quinto ed il sesto digits discriminano alleli che presentano delle mutazioni definite
sinonime in quanto consistono in una sostituzione di una singola base che non
comporta cambiamenti nell’espressione fenotipica della molecola.
Occasionalmente possono essere aggiunti un settimo ed un ottavo digits per
discriminare alleli che differiscono nella sequenza di introni o di regioni UTR (non
tradotte) all’estremità 5’ o 3’.
Al codice numerico possono essere aggiunti dei suffissi letterali che indicano
particolari caratteristiche di espressione dell’allele:
• L (low) una bassa espressione dell’allele sulla superficie cellulare.
• S (soluble) un allele che codifica per una proteina che è presente in forma
solubile e non associata alla membrana, es. le molecole HLA-G.
• C (cytoplasm) un allele il cui prodotto è ritrovabile nel citoplasma.
• A (aberrant) un allele la cui proteina non viene espressa o comunque porta
ad un’alterazione della struttura che non la rende funzionale.
• Q (questionable) un allele i cui effetti del prodotto genico non sono ancora
completamente definiti.
101.2 Correlazione tra alleli HLA e malattie
Uno degli sviluppi più interessanti in campo clinico degli studi sul sistema HLA a
livello di popolazione è nato dalla dimostrazione che molte malattie hanno una
associazione preferenziale con un determinato antigene o aplotipo.
Dalla prima osservazione, che risale al 1967 e che riguarda il morbo di Hodgkin, la
lista delle associazioni, nel tempo, si è notevolmente allungata e comprende
patologie molto differenti tra loro ma che presentano alcuni aspetti comuni.
In generale si tratta di malattie a patogenesi immunitaria che evidenziano: -frequente
ereditarietà, anche se la segregazione del carattere non segue le leggi mendeliane
perché a bassa penetranza (frequenza con cui il genotipo si esprime nel fenotipo) e
bassa espressività (grado di espressione fenotipica del genotipo), -decorso subacuto
o cronico, -abituale espressività dopo la maturità sessuale e scarso effetto sulla
riproduzione, -raramente influenzano la sopravvivenza in età fertile, -eziologia
multifattoriale nella quale hanno una forte rilevanza anche fattori extragenetici legati
all’ambiente.
Le informazioni sulle relazioni esistenti tra regione HLA e malattie sono state
raccolte attraverso sia “studi di popolazione” che “studi familiari”.
Nei primi viene paragonata la distribuzione degli antigeni HLA in gruppi di pazienti
e di soggetti controllo, omogenei da un punto di vista etnico, in quanto la frequenza
degli antigeni HLA varia notevolmente nelle diverse razze. Questi studi
evidenziano che la malattia tende ad esprimersi più facilmente se è presente il gene
che codifica un particolare antigene di istocompatibilità, quale che sia il motivo di
questa associazione.
11Nel caso particolare di una malattia che è condizionata da un gene, questo può
significare che il “gene malattia” è in linkage disequilibrium con un particolare gene
HLA. Molte sono le malattie che presentano questa correlazione.
Fig.4 Malattie associate con il sistema HLA
Come si può vedere dalla tabella, alcune malattie mostrano un’ associazione
preferenziale con un antigene codificato dal locus B, altre con quelli del locus C,
altre ancora con gli antigeni DR o DQ.
In alcuni casi, infine, sembra dimostrata un’associazione preferenziale con un intero
apoltipo HLA più che con un singolo allele. In alcuni casi, infine, l’associazione è
costante in tutte le popolazioni studiate ( ad esempio il B27 per la spondilite
anchilopoietica), altrove, è osservata solo in alcuni gruppi etnici.
Quindi è evidente ammettere l’intervento di un gene in linkage disequilibrium con
quel particolare antigene HLA in quella determinata popolazione e con specificità
diverse in gruppi etnici diversi.
Gli “studi familiari”, invece, dimostrano che il gene che determina la malattia viene
ereditato con alta probabilità insieme ad un gene che occupa un particolare locus
12nella regione HLA (gene che può variare da una famiglia all’altra); quindi questi
studi risultano utili per identificare la regione cromosomica del “gene malattia”, che
viene detto in linkage (cioè strettamente prossimo) con un particolare locus HLA,
indipendentemente dall’esistenza di un’associazione (che può esserci se esiste non
solo linkage, ma anche un linkage disequilibrium).
In questi casi, pertanto, diremo che quella “malattia” segrega insieme ad HLA.
Un esempio di questo tipo di patologie è dato dalla sindrome adrenogenitale
(deficienza della 21-OH-idrossilasi) che è una malattia monofattoriale recessiva in
cui il gene mutato CYP21 localizzato nella regione HLA di classe III è in linkage
disequilibrium con HLA-B47, situato a 681 Kb di distanza dal gene mutato.
Un altro esempio di malattie ereditarie è l’emocromatosi, in cui l’allele HLA-A03 è
in linkage disequilibrium con il gene mutato HFE, che dista 3697 kb dall’altro.
L’interesse della concatenazione fra il gene per una malattia ed il sistema HLA sta
nel fatto che essendo i prodotti HLA evidenziabili con tecniche di laboratorio
semplici, sia sierologiche che di biologia molecolare, nei familiari di persone affette
è possibile documentare la presenza dei geni per una determinata patologia anche
prima che questa si manifesti. Inoltre, per le malattie recessive, è anche possibile
identificare i portatori eterozigoti (“portatori sani”) del gene responsabile.
Un risultato comune emerso da entrambi gli studi è che la presenza di un
determinato antigene (o aplotipo) non conferisce automaticamente la malattia a tutti
gli individui che hanno quel particolare fenotipo, ma solo una suscettibilità sulla
quale agiscono altri fattori ereditari o ambientali. In altre parole l’individuo che ha
un certo antigene incorre, rispetto a chi non l’ha, in un rischio maggiore di
sviluppare la malattia ad esso associata. La misura di questo “rischio relativo”
13rappresenta un buon indice della forza di associazione e viene calcolata come rapporto tra la frequenza dell’antigene nei soggetti affetti dalla malattia e la frequenza dello stesso antigene nei sani. Un’associazione positiva è indicata da un rischio relativo >1 e negativa se
linfonodi regionali, queste cellule vengono “risvegliate” ed avviano quindi una
risposta autoimmune verso gli autoantigeni presentati dall’ APC.
In particolare è stato osservato che molte delle malattie associate agli antigeni DR
hanno una patogenesi immunitaria. Un esempio molto esplicativo è costituito dalle
malattie associate al fenotipo HLA-DR3 (il cui gene è in linkare disequilibrium con
HLA-B8). Come si può osservare si tratta di una serie di malattie d’organo
caratterizzate dalla produzione di autoanticorpi importanti nella patogenesi della
malattia : il diabete insulino dipendente, l’Addison idiopatico, ecc. E’ evidente
perciò che l’intervento del gene non consiste nel controllo della produzione di un
autoanticorpo particolare, ma che molto probabilmente il suo intervento si attua a
livello dei circuiti di controllo della produzione anticorpale, influenzando la
cooperazione tra linfociti T e linfociti B. Al contrario, il ruolo degli antigeni di
classe I nella risposta immune fa spiegare alcune delle associazioni trovate come
dovute all’intervento di geni la cui azione si esplicherebbe invece attraverso i T
citotossici.
Al di là dell’intervento dei geni della risposta immune vi sono altre ipotesi che, più o
meno indirettamente, prendono in considerazione il ruolo della regione HLA nella
risposta immune. Una di queste è “la teoria della mimesi” che ipotizza una possibile
somiglianza e quindi cross-reazione immunologica tra antigeni di istocompatibilità e
l’agente eziologico della malattia. Questa a sua volta aprirebbe due possibilità, sia
una mancata risposta immune e quindi una maggiore suscettibilità ad un agente
esogeno in alcuni casi che, una risposta cross-reagente e quindi un’autoreattività in
altri casi. Una variante di questa teoria è quella del “se stesso” modificato, che
prende in esame il ruolo degli antigeni HLA come molecole di presentazione
15dell’antigene virale, o batterico o parassitario al sistema immunitario. In questo
caso, però, l’autoaggressione sarebbe la conseguenza diretta di una reazione verso
strutture autologhe non più riconosciute come self. Pur considerando che la maggior
parte delle associazioni trovate può essere spiegata attraverso la modulazione della
risposta immune, vi sono alcune associazioni per le quali una simile spiegazione non
può essere chiamata in causa. Si pensa in questi casi che l’azione sia mediata
attraverso l’interferenza degli antigeni HLA, in quanto antigeni di membrana, con
l’azione di altri recettori e sistemi di membrana cellulare. Infatti, in alcuni casi, è
stata documentata tale interferenza, ad esempio quella esercitata dall’antigene B35
sul trasporto di magnesio intracellulare.
1.2.1 Principali malattie autoimmuni associate al sistema HLA
• SPONDILITE ANCHILOSANTE
La spondilite anchilosante è una malattia che fa parte del gruppo di artropatie
conosciute come spondiloartriti sieronegative, perché accumunate da alcune
caratteristiche quali: artrite periferica e sacroileite, lesioni cutanee, oculari (uveite
acuta anteriore) o genitali, assenza di fattore reumatoide, assenza di noduli
sottocutanei. Sintomo caratteristico è la perdita della mobilità spinale dovuta
all’infiammazione e/o al danneggiamento strutturale, causato dalla
osteoproliferazione. Non è del tutto chiaro se i due processi siano concatenati anche
se è noto che l’infiammazione porta alla deposizione di tessuto osseo. La causa della
spondilite anchilosante e delle altre spondiloartropatie è sconosciuta. Le principali
caratteristiche della malattia sono i desmofiti e la anchilosi che sono visibili, molti
mesi dopo se non anni, all’esame radiografico.
16Colpiscono principalmente il sesso maschile in età compresa tra la seconda e terza
decade; circa l’80% dei pazienti manifesta i primi sintomi di malattia al di sotto dei
30 anni e meno del 5% oltre i 45 anni.
L’associazione della malattia con l’allele HLA-B27, documentata oltre 25 anni fa,
resta la più classica associazione conosciuta con una forza di associazione elevata
che si avvicina al valore del 100%. Benchè il 95% circa dei pazienti affetti da
spondilite anchilosante sia portatore di questo antigene, è comunque necessario
precisare che questa malattia può insorgere anche in soggetti HLA-B27 negativi.
Se da una parte, quindi, l’antigene HLA-B27 rappresenta la componente genetica
più importante, dall’altra esso può non essere indispensabile, suggerendo
l’intervento di altri fattori genetici.
Studi epidemiologici hanno rilevato che la prevalenza di spondilite anchilosante
rispecchia la distribuzione dell’antigene HLA-B27 (ad esempio, la malattia è molto
rara nelle popolazioni dell’Africa Sub-Sahariana, dove HLA-B27 è virtualmente
assente).
Sono state proposte molte teorie per spiegare questa associazione, l’ipotesi più
accertata è data dalle proprietà biochimiche della molecola. L’HLA-B27 presenta
lentezza nel ripiegamento della catena pesante portando ad uno stress intracellulare
con conseguente attivazione di una risposta infiammatoria; inoltre la presenza di una
cys in posizione 67 della catena pesante contribuisce alla formazione di omodimeri
di catene pesanti in grado di legarsi a peptidi. L’espressione sulla superficie cellulare
di tali omodimeri mima le molecole di classe II attivando linfociti T CD4+
autoreattivi.
17Ad oggi sono conosciuti più di 43 sottotipi molecolari (alleli) della specificità HLA-
B27, codificante per il corrispettivo antigene. Gli alleli più comuni (HLA-B*27:05,
B*27:02, B*27:04, B*27:07) sono stati chiaramente associati con spondilite
anchilosante e con le altre spondiloartropatie in genere.
Mentre, due alleli, HLA-B*27:06 e B*27:09, caratteristici rispettivamente del sud-
est asiatico e della Sardegna non sembrano correlati con la malattia.
Entrambi questi alleli sono portatori di mutazioni che implicano una sostituzione
aminoacidica in posizione 114 e/o 116 della molecola: essendo localizzati sul
pavimento della tasca di legame al peptide, tali aminoacidi modificherebbero il
repertorio di peptidi che si possono legare ad HLA-B27, annullandone la sua
caratteristica di fattore di suscettibilità alle spondiloartropatie.
Il ruolo del laboratorio di tipizzazione HLA nella spondilite anchilosante è quello di
orientare un sospetto formulato precocemente, prima della comparsa del danno
osseo, in particolare è utilizzato un algoritmo diagnostico (“criteri di Berlino”) in cui
la presenza di dolore lombare cronico infiammatorio, caratteristiche cliniche ed
anamnestiche frequentemente presenti nelle spondiloartriti, le alterazioni dei
markers di flogosi sistemica, la risonanza magnetica nucleare e la positività per
HLA-B27 aumentano la specificità diagnostica.
Considerato che, dei numerosi alleli del gene HLA-B27, solo due sono riconosciuti
non essere correlati con la malattia (HLA-B*27:06 nel sud est asiatico e HLA-
B*27:09 in Sardegna), è di norma adeguato riconoscere la presenza dell’antigene o
del gene con test in bassa risoluzione. L’eventuale ricorso alla determinazione degli
alleli con test in alta risoluzione è da riservarsi ai soggetti originari delle popolazioni
sopra menzionate.
18• DIABETE DI TIPO 1
Il diabete mellito di tipo 1 (diabete insulino dipendente, IDDM della vecchia
classificazione) comprende, nei suoi due sottogruppi (idiopatico ed
immunomediato), le forme di malattia dipendenti, per il mantenimento della
omeostasi glucidica, dalla somministrazione esogena di insulina. Può insorgere a
qualsiasi età anche se generalmente si evidenzia entro i primi venti anni di vita.
E’ una malattia che si manifesta dopo un iniziale periodo silente e di lunghezza
molto variabile durante il quale vengono distrutte le β-cellule delle isole di
Langherans del pancreas, periodo seguito da disfunzioni e danni progressivi a lungo
termine a carico di svariati organi, come retina, reni, sistema nervoso e
cardiovascolare. Questa patologia è caratterizzata dalla produzione di autoanticorpi e
dalla infiltrazione di cellule del sistema immunitario nelle isole pancreatiche
(insulite), seguita dalla loro distruzione.
Studi condotti in modelli umani e murini hanno dimostrato che il processo
autodistruttivo è mediato da CD4, CD8 e macrofagi, che si accumulano in una
iniziale lesione delle isole. L’evento scatenante non è noto, anche se molto
probabilmente è causato da virus (ad esempio i citomegalovirus, i virus Coxackie B).
Una volta che le isole sono infiltrate da linfociti T e macrofagi si innesca una cascata
distruttiva, ovvero i macrofagi giocano il ruolo di APC presentando gli antigeni delle
cellule β ai linfociti CD4 e CD8, che producono contro tali cellule perforine e
citochine come Il-1, IL-2, IL-12, IL-17, IL-18, TNF-α. Inoltre i linfociti CD4+
attivano i linfociti B, che iniziano a produrre autoanticorpi. La maggior parte degli
autoantigeni noti sono associati a componenti delle cellule β, tra cui l’insulina.
Quest’ultima è il primo antigene target rintracciabile durante la progressione iniziale
19del diabete, successivamente si ha un aumento del numero di autoantigeni e di
epitopi degli autoantigeni che va di pari passo con la progressione e l’aumento di
severità della malattia. Le prime manifestazioni cliniche del diabete in un individuo
si manifestano dopo che il 90% delle isole è ormai distrutto.
Lo studio della familiarità e delle modalità di trasmissione della malattia ha
dimostrato come questa patologia sia correlata a fattori ambientali ed individuali
caratterizzati da una predisposizione di tipo multigenica.
Componente essenziale di questo background genetico è il sistema HLA.
L’incidenza della malattia presenta un gradiente decrescente nord-sud con un picco
d’incidenza nel Nord-Europa. La malattia presenta una rilevante familiarità con una
concordanza del 30-50% nelle coppie di gemelli monovulari mentre il rischio di
ammalarsi per un fratello di un soggetto affetto da IDDM è di circa il 6%.
E’ inoltre emersa una maggiore incidenza di soggetti affetti fra i figli di uomini
ammalati rispetto a donne affette da IDDM; infine la possibilità che una bambina
affetta da IDDM avesse un padre ammalato era maggiore di quella di un figlio
maschio mentre la relazione reciproca tra madre e figlio non è significativa.
Dagli anni ’80 in poi gli alleli DR3-DR4 sono considerati marcatori privilegiati della
suscettibilità alla malattia con effetti sinergici legati alla contemporanea espressione
dei due alleli nello stesso individuo.
Studiando una popolazione di soggetti affetti da IDDM ed eterozigoti DR3/DR4 fu
dimostrato che la suscettibilità era legata agli alleli dei geni HLA-DQA1 e HLA-
DQB1 che codificano per molecole in grado di formare eterodimeri estremamente
polimorfi. Si vide inoltre che gli alleli del locus HLA-DQB1, oltre alla
predisposizione all’IDDM erano associati alla resistenza; in particolare, gli alleli non
20presentanti un residuo di aspargina in posizione 57 della sequenza aminoacidica
della catena DQβ conferivano resistenza alla malattia (HLA-DQB1*03:02), mentre
la presenza di un residuo di arginina in posizione 52 della catena DQα era correlata
con un certo grado di suscettibilità alla malattia.
In conclusione, al fine di ottenere una corretta identificazione degli aplotipi
predisponenti, in laboratorio, la tipizzazioni principale da eseguire è l’ HLA-DRB in
bassa risoluzione.
• ARTRITE REUMATOIDE
L’artrite reumatoide, tra tutte le malattie associate al sistema HLA, è quella con la
più alta prevalenza, pari all’1%. E’ una poliartrite infiammatoria cronica con
manifestazioni sistemiche (noduli reumatici, vasculite, impegno oculare e viscerale,
neuropatia), a genesi autoimmune, caratterizzata da localizzazione primitiva del
processo patologico nella membrana sinoviale delle articolazioni diartrodiali, delle
guaine tendinee e delle borse. Essa è a carattere erosivo, ad andamento progressivo,
con evoluzione in dislocazione e distruzione completa della cartilagine articolare ed
esito finale in anchilosi.
E’ più frequente nella donna che nell’uomo, solitamente esordisce nella quinta
decade anche se può comparire in tutto il corso dell’età adulta; esistono inoltre
alcune forme di artrite reumatoide tipiche dell’età infantile, alcune delle quali sono
solo l’esordio precoce dell’artrite reumatoide dell’adulto.
Analogamente ad altre malattie autoimmuni, l’eziologia dell’artrite reumatoide è
complessa e multifattoriale; sia fattori genetici che ambientali influenzano la
suscettibilità ad ammalarsi, la gravità, la prognosi nonché la risposta alla terapia.
21La diagnosi di artrite reumatoide si basa sulla presenza di un certo numero di criteri
clinici, non particolarmente specifici per la malattia. Uno dei criteri è la presenza del
fattore reumatoide (FR), un autoanticorpo diretto alla porzione Fc delle IgG che ha
una sensibilità del 60-70% ed una specificità del 80-90%.
Inoltre è rilevante la presenza di anticorpi contro peptidi ciclici citrullinati (anti-
CCP) associati al danno articolare; la sensibilità di questo esame è simile a quella
del FR ma la specificità è significativamente superiore (99%). Entrambi questi
autoanticorpi possono essere presenti anche per diversi anni prima della comparsa
della malattia e sono associati alla gravità della malattia stessa.
L’importanza del fattore genetico è emersa dai primi lavori di Peter Stratsny nel
1976, dove è stata evidenziata l’associazione tra HLA e artrite reumatoide e da
allora molti immunogenetisti si sono occupati di questo argomento.
E’ stato visto che la suscettibilità ad ammalarsi di artrite reumatoide è fortemente
associata al gene HLA-DRB1; inoltre, in particolare sono interessati gli alleli del
gruppo HLA-DRB1*04 (HLA-DRB1*04:01, *04:04, *04:05, *04:08) e HLA-
DRB1*01:01, *01:02, *10:01, *14:02.
Un’analisi dei prodotti di questi geni dimostra che condividono una sequenza di 5
aminoacidi nelle posizioni 70-74 della terza regione ipervariabile della molecola
HLA-DRB1. Questi aminoacidi sono essenziali per la conformazione della tasca 4,
in particolare gli aminoacidi in posizione 71, che presentano carica positiva e
quindi consentono il legame solo con residui con carica negativa. Questa
osservazione ha gettato le basi dell’ipotesi della “shared epitope” (motivo
condiviso). Questo modello evidenzia che questi aminoacidi predispongono
all’artrite reumatoide influenzando la presentazione dell’antigene nella tasca oppure
22agendo come peptidi self in grado di modulare l’educazione timica dei T linfociti.
Nel processo di educazione timica, potranno essere selezionati, perché a bassa
affinità, cloni autoreattivi per la suddetta sequenza (come per altri epitopi del self)
che potranno espandersi in un secondo momento in seguito a infezione specifica e
poi migrare nelle sinovie, dove possono essere “intrappolati” per l’azione di
citochine.
Una seconda ipotesi sostiene che, sempre nella stessa regione della molecola DRB1
(aminoacidi 70-74), esistono sequenze che conferiscono protezione nei confronti
dell’artrite reumatoide (rheumatoid arthritis protection); in questo caso, l’importanza
della sequenza 70-74 non sarebbe tanto nella modificazione della tasca 4 quanto nel
peptide che potrebbe legarsi a molecole HLA-DQ ed essere riconosciuto da cellule
T regolatorie che inibirebbero i linfociti autoreattivi.
In particolare alcuni autori hanno studiato la correlazione tra alleli associati con
suscettibilità e quelli associati a protezione con anticorpi anti-CCP ed hanno
evidenziato che tutti i pazienti con anticorpi anti-CCP avevano gli alleli associati
con suscettibilità, mentre erano assenti nei pazienti senza anticorpi anti-CCP, mentre
per i pazienti con alleli associati a protezione, la protezione era indipendente dalla
presenza o meno di anticorpi anti-CCP. Quindi usando l’HLA e gli anticorpi anti-
CCP è possibile suddividere l’artrite reumatoide in “sottotipi” e prevederne la
prognosi.
Inoltre, con tecniche di biologia molecolare, è emerso che l’ allele HLA-
DRB1*04:01 ed in minor misura l’allele HLA-B1*04:03 sono coinvolti non solo
nella suscettibilità alla malattia ma rappresentano un marker prognostico sfavorevole
( i soggetti positivi presentano un fenotipo clinico più grave) e l’allele HLA-
23DBR1*04:02 è perfino coinvolto nella resistenza alla malattia, i soggetti positivi
presentano una minore incidenza alla malattia.
• CELIACHIA
La malattia celiaca è un’enteropatia indotta dall’intolleranza al glutine, nei confronti
del quale si stabilisce una reazione immune che provoca danni alla mucosa
intestinale del digiuno con conseguente atrofia dei villi, iperplasia delle cellule delle
cripte ed infiltrazione di cellule linfoidi.
Nella forma classica si presenta nei primi 6-24 mesi di vita ed è caratterizzata da
diarrea cronica, distensione addominale e scarsa crescita. La forma atipica, invece, si
presenta nell’adulto con manifestazioni prevalentemente extraintestinali quali
anemia, osteoporosi, alopecia, infertilità. E’ nota anche una forma silente, del tutto
asintomatica nonostante la presenza dell’atrofia della mucosa intestinale, e una
forma latente dove la mucosa risulta normale ma sono presenti gli anticorpi
caratteristici della malattia.
La celiachia è stata perciò paragonata ad un iceberg la cui porzione emergente
rappresenta le forme clinicamente manifeste (tipica o atipica), mentre la parte
sommersa, di maggiori dimensioni, comprende i casi silenti e latenti.
Questa ampia variabilità clinica e la conseguente difficoltà diagnostica hanno reso
necessari anni per definire correttamente l’epidemiologia della malattia.
Infatti, parallelamente alla disponibilità dei test diagnostici sempre più sensibili, la
stima della prevalenza della celiachia è andata aumentando nel tempo.
Ad oggi, l’incidenza in Italia è 1/150 persone. Colpisce più spesso il sesso
femminile con un rapporto di 2 femmine/1 maschio.
24La mancata o tardiva diagnosi o la non aderenza alla dieta espone i pazienti al
rischio di sviluppare malattie di vario tipo, in primo luogo altre patologie
autoimmuni, ed insorgenza di neoplasie dell’apparato gastroenterico quali il linfoma
non Hodgkin e varie forme di neoplasia epiteliale.
Una prevalenza più elevata di quella della popolazione generale è stata riscontrata in
numerosi gruppi a rischio che comprendono familiari di celiaci, in particolare di
primo grado, soggetti con sindrome di Down, Turner, Williams e pazienti con altre
malattie autoimmuni quali diabete mellito insulino-dipendente, epatite autoimmune
e sindrome di Sjogren. L’associazione tra la malattia celiaca ed altre patologie
autoimmuni è considerata almeno in parte dovuta ad un comune background
genetico. L’importanza di diagnosticare queste forme di celiachia associate a
patologia autoimmune è duplice, dal momento che la dieta aglutinata non è in grado
di prevenire le manifestazioni cliniche dell’enteropatia da glutine, ma anche di
determinare un miglioramento del quadro clinico almeno per alcune delle patologie
associate.
Numerosi studi riportano che la malattia celiaca si associa frequentemente alla
presenza di specifici geni del sistema HLA, codificanti gli eterodimeri DQ2 e DQ8,
identificabili tramite gli alleli DQA1*05:01/DQB1*02:01 o
DQA1*05:01/DQB1*02:02 e DQB1*03:02 rispettivamente.
Poiché gli alleli DQB1*02:01 e DQB1*02:02 sono identici eccetto che per
l’aminoacido 135 localizzato nel dominio più prossimo alla membrana della catena
DQβ che non influenza la sequenza delle tasche di presentazione della molecola,
appare evidente che la presenza sulla membrana di eterodimeri
DQA1*05:01/DQB1*02:01 o DQA1*05:01/DQB1*02:02 attribuisce alle cellule
25che li espone sulla membrana la stessa efficacia nel presentare i peptidi della
gliadina ai linfociti T. Quest’ultimi, isolati dalle biopsie dell’intestino tenue di
pazienti celiaci, riconoscono preferenzialmente peptidi deaminati, che sono prodotti
tramite l’azione dell’enzima transglutaminasi (tTG), che catalizza la trasformazione
della glutammina in acido glutammico, carico negativamente; l’acquisizione di
cariche negative da parte dei peptidi della gliadina consente una maggiore affinità
con le molecole HLA-DQ2 e DQ-8 sulle cellule presentanti l’antigene (APC).
Si ha l’attivazione dei linfociti T-helper, che producono sia un gran numero di
citochine inducenti una notevole risposta infiammatoria nella mucosa e nella lamina
sottomucosa del digiuno, che l’attivazione di linfociti B in grado di produrre
anticorpi specifici anti-gliadina, anti-endomisio e anti-tTG. Quest’ultimi attivano
una risposta immunitaria distruttiva verso i tessuti che la contengono: il più colpito
è la mucosa intestinale, ma il danno riguarda organi diversi (sistema nervoso, cute,
pancreas, fegato).
L’aplotipo DQ2 è stato osservato nel 90-95% dei pazienti, mentre l’aplotipo DQ8 è
presente in circa il 5%. Esiste altresì una quota di pazienti celiaci (meno del 2%)
che non possiede né il DQ2 né il DQ8, così come è necessario considerare che circa
il 25-30% della popolazione presenta i suddetti aplotipi predisponesti senza mai
sviluppare la malattia.
Si deduce pertanto che la ricerca degli aplotipi DQ2 e DQ8 presenta un valore
predittivo negativo molto forte ed un debole significato predittivo positivo per la
diagnosi di malattia celiaca. La presenza di questi alleli, pur essendo un importante
fattore di predisposizione genetica, ovviamente non può essere considerato l’unica
26causa della malattia, ma altri fattori ambientali e genetici influiscono sulla
patogenesi.
La diagnosi viene effettuata avvalendosi dell’utilizzo di linee-guida approvate dal
Comitato Nazionale per la sicurezza Alimentare (pubblicato in G.U.n°.32 S.O. del 7
febbraio 2008), che consentono di disporre di protocolli semplici, applicabili su tutto
il territorio nazionale ed in grado di identificare il maggior numero di celiaci e di
assicurarne il monitoraggio. Vengono utilizzati tre diversi percorsi diagnostici in
base alla presenza di pazienti con forte sospetto clinico di celiachia
(malassorbimento franco, caratterizzato da significativo calo ponderale, diarrea ed
astenia severa), o con bassa probabilità di celiachia (casi mono o paucisintomatici)
oppure a genitori e fratelli di celiaci.
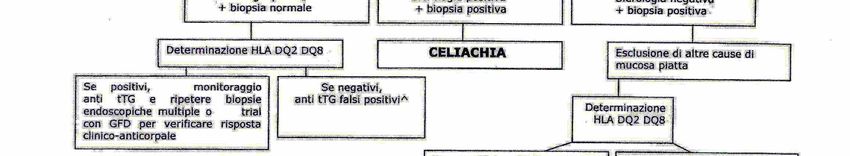
Come si può osservare dalle tabelle sottostanti i saggi di primo livello sono
rappresentati dalla biopsia duodenale e da markers anticorpali (IgA sieriche e anti-
tTG), mentre l’HLA, come test di secondo livello, deve essere eseguito quando i
primi non hanno portato ad una diagnosi certa.
Per la tipizzazione HLA in prima istanza viene effettuata la ricerca dell’eventuale
presenza dell’aplotipo HLA-DQ2, HLA-DQ8 o entrambi in biologia molecolare in
basso livello di risoluzione ed, in caso di positività, l’espressione degli alleli sia
DQB che DQA in alta risoluzione. Con l’alta risoluzione, a seconda dell’allele
espresso si identifica il “gruppo di rischio”, questo è rilevante, ad esempio, per i
familiari di primo grado dell’affetto, in quanto consente di stabilire la frequenza
degli esami sierologici da effettuare nel tempo per seguire con massima attenzione
la possibile insorgenza della malattia.
27Fig.5 Percorso diagnostico per pazienti con elevato sospetto di celiachia
Fig.6 Percorso diagnostico per pazienti con basso sospetto di celiachia
28Fig. 7 Percorso diagnostico per familiari di I grado
• MALATTIA BEHCET
La malattia di Behçet è una patologia sistemica di tipo infiammatorio ad
eziopatogenesi sconosciuta. E’ caratterizzata dalla triade clinica: aftosi orale, aftosi
genitale (recidivanti) ed infiammazione oculare. Può tuttavia frequentemente
coinvolgere anche articolazioni, cute, sistema nervoso centrale, apparato
gastroenterico e vasi sanguigni. Classificata tra le vasculiti sistemiche, può
coinvolgere sia i vasi venosi che arteriosi di qualsiasi calibro e distretto corporeo,
determinando quindi differenti presentazioni cliniche.
La malattia ha un decorso cronico recidevante, la cui espressione clinica e gravità
sono estremamente variabili da paziente a paziente, anche in base alla provenienza
geografica; il coinvolgimento d’organo risulta infatti più severo nelle aree asiatiche
rispetto a quelle occidentali. In particolare risulta endemica in aree quali Turchia,
29Iraq, Iran, Corea e Giappone, è meno comune nell’Europa settentrionale e nel Nord
America e rara nelle popolazioni africane.
Colpisce prevalentemente giovani adulti soprattutto di sesso maschile tra la seconda
e quarta decade di vita, anche se possono essere presenti casi nelle fasce sia senili
che infantili.
L’eziologia della malattia di Behçet non è nota; da diversi studi emerge che la causa
potrebbe essere un evento infettivo che, tramite meccanismi di mimesi antigenica
con antigeni-self, induce attivazione e cronicizzazione di un abnorme risposta
immunitaria contro un autoantigene.
Gli autoantigeni candidati sono l’antigene S retinico, le proteine HSP e l’α-enolasi
riconosciute dagli anticorpi frequentemente riscontrati in corso di malattia, seppur
non specifici.
Altri studi hanno inoltre documentato un ruolo significativo di una aberrante
generalizzata risposta T cellulare. Infatti rispetto ai controlli sani, i pazienti affetti
presentano un aumentato numero di linfociti T CD4+ circolanti, il cui target
antigenico non è noto, ma esprimono markers precoci di attivazione e producono
citochine infiammatorie, tra INF- gamma e TNF-α.
Questo potrebbe essere dovuto ad un difetto di trasduzione del segnale da parte dei
linfociti T, i quali presenterebbero una più bassa soglia di attivazione da parte di
multipli stimoli antigenici.
La diagnosi si basa esclusivamente su elementi clinici, anche se l’associazione con
l’HLA fornisce un supporto allo specialista. Infatti, dal 1973 è stata evidenziato che
l’eterodimero HLA-B51 ha un ruolo primario nella suscettibilità alla malattia, in
30particolare il 20% degli individui sani appartenenti a diverse origini etniche sono
HLA-B1 positivi, mentre la percentuale di positività passa dal 50 all’80% nei malati.
Non è tuttavia conosciuto il meccanismo molecolare mediante il quale la molecola
HLA-B51 conferisce un’alta predisposizione alla malattia.
Da studi di eluizione dei peptidi ancorati a molecole B51 è emerso che la tasca del
sito di legame della molecola HLA-B51 alloggia con scarsa affinità i peptidi
determinando un difetto di presentazione all’azione citotossica dei linfociti con
possibilità di riattivazione dopo una nuova infezione non nota.
Allo stato attuale delle conoscenze scientifiche, di fronte ad un sospetto di malattia
di Behçet, viene effettuata la tipizzazione del locus HLA-B in sierologia o in
biologia molecolare a basso livello di risoluzione.
1.2.2. Principali malattie infettive associate al sistema HLA
Le malattie infettive strettamente associate con il sistema HLA sono poche.
Questo dato è causato sia dalla molteplicità dei determinati antigenici microbici che
dalla risposta immune di ogni individuo nella quale sono coinvolti loci HLA e non-
HLA che possono mascherare l’effetto dei primi.
Inoltre il numero dei soggetti studiati è spesso insufficiente per raggiungere una
significatività statistica e a questo consegue che l’ampiezza dei campioni in studio
non consente di individuare associazioni alleliche più forti di un Odd Ratio compreso
tra 0,5 e 2.
Per lo studio delle malattie infettive, infatti, si preferisce analizzare degli alberi
genealogici più semplici, ovvero solo due o più fratelli affetti con i genitori in modo
31da evitare il problema di classificare membri della famiglia che potrebbero non
essere stati esposti all’agente infettivo.
Mentre gli studi di associazione vengono effettuati sulle popolazioni in cui la
malattia studiata è endemica.
Da numerose ricerche sono stati individuati alcuni fattori di suscettibilità o resistenza
genetica sia a parassitosi, che infezioni batteriche che virali.
Ad esempio, da studi di associazione, sono stati identificati i geni di resistenza alla
forme gravi di malaria in popolazioni dell’Africa Sub-Sahariana. E’ stato dimostrato
che l’allele HLA-B53 riduce il rischio di morte per malaria grave indotta dal
Plasmodio Falciparum di circa il 40%. Questo allele ha una frequenza molto bassa
nelle popolazioni non africane, è presente invece nel 25% dei soggetti sani e nei
bambini affetti da forme lievi di malaria nella popolazione africana. Questo dato è
presumibilmente correlato all’efficienza da parte dell’allele HLA-B53 a presentare i
peptidi derivati dagli sporozoiti e questo induce la formazione di cloni di linfociti T
CD8+ molto efficienti nell’eliminazione del parassita nello stadio di infezione
epatica. In effetti, tali CD8 sono stati riscontrati nei pazienti affetti da malaria e
peptidi sporozoitici sono stati eluiti dalle molecole HLA-B53 di questi pazienti.
L’incremento della frequenza dell’allele HLA-B53 in queste popolazioni rispetto alla
frequenza riscontrabile in altre popolazioni non selezionate dalla malaria, suggerisce
che questo incremento sia il risultato di un’elevata pressione selettiva esercitata dal
patogeno.
Sono state evidenziate associazioni con il sistema HLA anche per infezioni
batteriche, ad esempio in Asia il fenotipo HLA-DR2 è correlato con la suscettibilità
sia alla lebbra che alla tubercolosi.
32Per ciò che concerne l’infezione da HIV, invece, nonostante l’enorme mole di studi
condotti sull’argomento, non è emersa nessuna associazione consistente con
particolari alleli di classe I e II. L’inconsistenza di queste associazioni potrebbe
essere legata sia alla presenza di estensivi polimorfismi nel virus che dall’alto tasso
di diversificazione dell’ospite. Sono state trovate, però, diverse associazioni,
confermate in più di uno studio, con l’andamento della malattia. E’ stato dimostrato
che l’allele HLA-B53 è associato con una progressione più rapida della malattia,
mentre il fenotipo HLA-B27 con una progressione più lenta.
Infine diversi studi hanno dimostrato che la negatività per l’allele HLA-B*57:01 è
correlata con la mancata insorgenza della reazione di ipersensibilità al trattamento
terapeutico con abacavir. Quest’ultimo è un farmaco che, nel 5-8 % dei pazienti
causa una reazione clinica indesiderata che consiste nell’insorgenza di una sindrome
clinica multiorgano caratterizzata da febbre, rash cutaneo, cefalea e disturbi
gastrointestinali che si osservano generalmente entro sei settimane dall’inizio del
trattamento. Il risultato del test genetico, quindi, è di notevole importanza, però non
deve sostituire un accurato counselling sulle possibili reazioni avverse e un’attenta
osservazione clinica, dal momento che un risultato negativo non esclude in assoluto
la possibilità che l’evento avverso si manifesti.
Interessante per i risvolti clinici è poi l’associazione dell’allele HLA-DR7 con
l’infezione da citomegalovirus nei pazienti sottoposti a trapianto renale e nei pazienti
affetti da AIDS; inoltre questo allele è un fattore di suscettibilità alla cronicizzazione
delle infezioni virali di epatite B e C. Quindi questo allele presenta un’incapacità a
presentare efficacemente gli antigeni virali.
33In conclusione, questi studi presentano un profondo significato biologico ma un
indubbio risvolto clinico. L’eventuale individuazione di alleli con un elevato rischio
di suscettibilità ha infatti un valore sia nel delucidare i meccanismi patogenetici della
malattia che nel sottoporre gli individui a rischio a un’adeguata profilassi.
1.3 SCOPO DELLA TESI
Nel corso del presente lavoro di tesi, svolto presso il laboratorio di tipizzazione
tissutale HLA dell’AUSL 6 di Livorno, sono stati esaminati, nell’anno 2013, i
pazienti richiedenti un’eventuale predisposizione genetica per patologie associate al
sistema HLA, in particolare per la spondilite anchilosante, per la malattia celiaca, per
il diabete mellito di tipo I, per la malattia di Behçet e per valutare nei pazienti hiv
positivi l’eventuale insorgenza della reazione di ipersensibilità al trattamento con
abacavir.
La presenza dell’antigene HLA-B27 per la spondilite anchilosante e dell’antigene
HLA-B51 per la malattia di Behçet è stata evidenziata con il test di
microlinfocitotossicità complemento dipendente (tecnica sierologica), che valuta la
percentuale di cellule positive ottenute tramite la citotossicità mediata dal
complemento che danneggia le cellule riconosciute dall’anticorpo specifico, secondo
score internazionali standardadizzati
La tipizzazione HLA per la malattia celiaca, per il diabete mellito di tipo I e per la
ricerca dell’allele HLA- B*57.01 nei pazienti HIV positivi è stato effettuata con la
PCR-SSP (test di biologia molecolare), metodica che consente di riconoscere un
allele o un gruppo di alleli con l’impiego di primer sequenza specifica.
34Come possiamo osservare dalla tabella sottostante i pazienti analizzati, suddivisi in
richieste esterne e dai reparti ospedalieri, sono risultati 376, di cui:
• 119 per la spondilite anchilosante (16 interni/103 esterni),
• 27 per la malattia di Behçet (7 interni/20 esterni),
• 145 per la malattia celiaca (3 interni/142 esterni),
• 30 per il diabete di tipo I (12 interni/18 esterni),
• 55 per i pazienti HIV positivi (27 interni/28 esterni).
Patologie associate sistema HLA Allele/Aplotipo Interni Esterni
SPONDILITE ANCHILOSANTE B27 16 103
MALATTIA BEHCET B51 7 20
MALATTIA CELIACA DQ2/DQ8 3 142
DIABETE TIPO I DR3/DR4 12 18
HIV: TERAPIA ABACAVIR B*57:01 27 28
Tab. 3 Pazienti analizzati anno 2013 AUSL 6 di Livorno
35Puoi anche leggere

















































