MMMM Progettazione di nuovi farmaci mediante tecniche computazionali
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
G
M
M
Progettazione di nuovi farmaci mediante
tecniche computazionali
In silico techniques for drug design
Ferdinando Spagnolo *
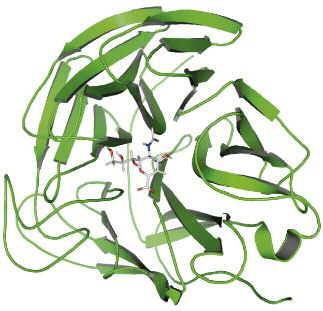
Fig. 1 - Anticorpo monoclonale anti VEGFR-1 progettato con metodologie di homology modeling. (Monoclonal antibody
against VEGFR-1 designed with homology modeling methodologies) Immagine F. Spagnolo.
Riassunto - L’uso di strumenti computazionali per la progettazione dei farmaci è diventato quotidiano ed assolutamente necessario al fine di
ottimizzare tutti i passaggi che portano alla realizzazione di un nuovo farmaco. Infatti, le tecnologie computazionali consentono di costruire
simulazioni capaci di integrare tutte le informazioni ottenute con le tecniche sperimentali tradizionali ed ottenere modelli virtuali assolutamente
aderenti ai fenomeni chimici, fisici e biologici coinvolti nell’interazione farmaco-recettore. Oltre agli evidenti vantaggi connessi con il risparmio
di tempo e mate prime, gli esperimenti condotti in silico consentono una riproducibilità e precisione assolute, abbattendo il rischio connesso
con la manipolazione di reagenti potenzialmente dannosi all’ambiente. La virtualizzazione consente molteplici operazioni che vanno dalla
visualizzazione di molecole alla simulazione di reazioni chimiche che avvengono nell’organismo vivente nei siti d’azione dei farmaci. Pertanto
il drug design consente una maggiore esplorazione dello spazio chimico che permette di guidare gli scienziati nel disegno, progettazione e
caratterizzazione di nuovi farmaci o trovare nuovi impieghi farmaceutici di molecole già note. Di seguito verranno descritti alcuni successi e
metodi computazionali nello sviluppo di nuovi farmaci.
Parole chiave: dinamica molecolare, progettazione farmaci, docking, calcoli ab intio.
Summary - The use of computational tools for drug design become daily and necessary in order to optimize all of the steps that lead to the
realization of a new drug. In fact, the computational technologies allow the generation of simulations that can integrate all the information
obtained with the experimental techniques and traditional virtual models get absolutely adhering to the chemical phenomena, physical and
biological factors involved in the drug-receptor interaction. In addition to the obvious benefits associated with time-row-material-saving, in
silico experiments allow better reproducibility and pinpoint accuracy, reducing the risk associated with the handling of reagents potentially
harmful to the environment. Virtualization allows multiple operations ranging from visualization of molecules to the simulation of chemical
reactions that occur in the living organism in the sites of action of drugs. Therefore, the drug design allows for greater exploration of the
chemical space that can guide scientists in design, design and characterization of new drugs or find new uses of pharmaceutical molecules
already known. Below are some of the successes and computational methods in the development of new drugs.
Key words: drug design, molecular dynamics, docking, ab initio calculations.
* Cap. sa. (farm.) Centro Studi e Ricerche di Sanità e Veterinaria dell’Esercito, Roma.
Scuola di Dottorato Scienze Biomediche e Biotecnologiche – Bioinformatica - Università di Udine.
G Med Mil. 2013; 163(1): 31-48 31
G
M
M
Introduzione
Già i primi calcolatori costruiti
negli anni ’40 vennero utilizzati per la
descrizione di sistemi atomici e mole-
colari. Nell’ultimo ventennio del XX
secolo le tecniche computazionali
hanno raggiunto la piena maturità
nell’applicazione dei sistemi chimici,
fisici, biologici e chimico-farmaceutici.
In questi anni i metodi IT (Informa-
tion Technology) hanno affiancato le
tecniche tradizionali utilizzate in labo-
ratorio sia nella progettazione di nuovi
farmaci, sia nell’ottimizzazione di
farmaci già esistenti. Tra i successi,
ormai diventati classici esempi di
farmaci progettati con l’ausilio di Fig. 2 - Visualizzazione 3D delle strutture HSG1.pdb ed indinavir.
tecniche in silico, possiamo citare la La struttura HSG1 è il modello tridimensionale dell’HIV-1 polimerasi. In figura, il
dimero è rappresentato in cartoon azzurro, catena A, e verde, catena B. Il sito
Norfloxacina (1), capostipite dei fluo- attivo dell’enzima è posto all’interfaccia del dimero. In questo modello l’indinavir si
rochinoloni, progettato negli anni ’80, posiziona nel sito attivo inibendo irreversibilmente il dimero. Immagine F. Spagnolo
una delle più importanti famiglie di
antibatterici che vede, tra gli altri, la
Ciprofloxacina uno dei più importanti mente preziosi contributi per lo Razionale del drug design
derivati. Pochi anni più tardi, nel 1994, sviluppo di farmaci “orfani” ossia quei
veniva progettato l’Indinavir, inibitore farmaci utili alla cura di patologie La scoperta e lo sviluppo di nuovi
di un enzima chiave per la vitalità del molto rare che, dato il basso impatto farmaci è un intenso sforzo interdisci-
virus dell’immunodeficienza acquisita economico, non sono al centro degli plinare. Generalmente la scoperta di
di tipo I (HIV-I), rappresentato con il interessi di ricerca e sviluppo dell’in- nuovi farmaci è raffigurata come un
suo recettore, HIV-1 polimerasi, in dustria farmaceutica. Le prestazioni processo lineare, consecutivo, che inizia
figura 2, e nel 1997 un suo derivato dei moderni calcolatori e l’accessibi- con la scoperta del bersaglio biologico
di seconda generazione, il Nelfinavir lità delle risorse informatiche hanno e del composto lead (molecola dotata
(1997) (2), utilizzando il docking, una permesso, tra l’altro, di avviare di una specifica grossolana attività biolo-
delle tecniche computazionali più progetti no-profit su scala internazio- gica e che può essere ulteriormente
avveniristiche dell’epoca. Oramai, nale per lo sviluppo di nuovi farmaci modificata al fine di modularne gli effetti
nello scenario della chimica farmaceu- contro la malaria, tubercolosi, leish- biologici). L’ottimizzazione del lead
tica, non esiste oggi esempio di maniosi ed AIDS (e.g. progetto fight procede fino alla fase pre-clinica in vitro
farmaco già sviluppato, od in corso di AIDS@home (3) e GO Fight Against ed in vivo per determinare se i composti
sviluppo, che non abbia risentito, in Malaria (4)…). I limiti principali del elaborati soddisfano una serie di criteri
almeno una fase del suo percorso, computational aided drug design pre-impostati per l’avvio dello sviluppo
dell’influenza di almeno una metodo- (CADD) risiedono nell’intrinseca diffi- clinico. L’esigenza dell’ottimizzazione
logia computazionale. La razionalizza- coltà di elaborare modelli matematici delle risorse per lo sviluppo di un nuovo
zione dei costi e l’ottimizzazione del sufficientemente completi per rappre- farmaco è implicita quando si analiz-
processo di sviluppo che caratteriz- sentare in maniera integrale i sistemi zano i costi, i tempi e la richiesta del
zano le tecniche chimico-farmaceu- biologici complessi alla base della farmaco risolutivo per una patologia
tico-computazionali, offrono certa- fisiologia cellulare. specifica. Infatti il numero di anni per
32 G Med Mil. 2013; 163(1): 31-48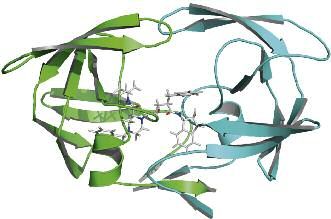
G
M
M
portare un farmaco dalla scoperta al
mercato è di circa 12-14 anni ed hanno
un costano indicativo compreso tra $ 1,2
e $ 1,4 miliardi di dollari (5) (Fig. 3).
Tradizionalmente, i farmaci venivano
scoperti sintetizzando composti in
processi multi-step dove i principi attivi
venivano testati su sistemi biologici sele-
zionati, prima in vitro, successivamente
in vivo, per selezionare ulteriormente i
candidati lead più promettenti per le
loro proprietà farmacocinetiche, meta-
bolismo e tossicità potenziale. Un tale
processo di sviluppo è caratterizzato da
alti tassi di abbandono con insuccessi
attribuibili a scarsa farmacocinetica,
mancanza di efficacia, tossicità animale,
Fig. 3 - Fasi dello sviluppo di un nuovo farmaco: dei 5,000 composti candidati
effetti indesiderati nell’uomo e vari
a diventare farmaco, mediamente, solo uno è immesso in commercio. L’utilità
fattori commerciali. Oggi, il processo di del drug design si percepisce già nello studio preliminare dei composti
scoperta di nuovi farmaci è stato rivo- candidati lead in modo che solo per una frazione delle centinaia di migliaia di
luzionato con l’avvento della genomica, composti venga proposta una sintesi, continua poi conferendo strumenti
analitici per l’analisi dei risultati dei test preclinici e, oggigiorno anche clinici.
proteomica, bioinformatica e tecnologie
altamente efficienti quali, la chimica
combinatoriale, high throughput scree- cidazione riguardo le loro funzioni, alla conformeri, suggerendone potenziali
ning (HTS), screening virtuale, proget- scoperta e sviluppo di leads con le ligandi farmacologicamente attivi
tazione de novo, ADMET (Assorbimento, proprietà desiderate. I ruoli principali (chiavi). La struttura risolta di un
Distribuzione, Metabolismo, Elimina- del calcolo scientifico nella scoperta di complesso ligando-recettore fornisce
zione, Trasformazione) screening in un principio attivo sono: una visione dettagliata delle interazioni
vitro ed in silico. I metodi virtuali 1 Screening virtuale e progettazione tra il ligando e lo stesso recettore. Lo
possono aiutare ad identificare i bersagli de novo; studio minuzioso di un composto con
del principio attivo oltre che descriverne 2 Previsione in silico ADME / T; le caratteristiche drug like consente di
le proprietà chimico-fisiche. Gli stru- 3 Caratterizzazione dell’interazione proporre modifiche strutturali capaci di
menti comuni a tutti i metodi computa- recettore/ligando. modulate la formazione del complesso
zionali sono sistemi di gestione dei dati Con queste premesse, uno dei prin- ligando-recettore del composto modifi-
(database ottimizzati), calcolo scientifico cipi cardine per il moderno computa- candone l’ADME.
ad alte prestazioni (High Performance tional drug design, rimane, in ogni caso, Un composto drug like (o druggable)
Computing - HPC) ed internet. L’accesso il modello chiave-serratura (6), elabo- contiene gruppi funzionali e/o possiede
ad una inimmaginabile quantità di infor- rato poi come modello dell’adattamento proprietà chimico fisiche in linea con la
mazioni e la trasformazione dei dati indotto (7). La biologia molecolare, maggior parte dei farmaci noti. Strutture
biologici complessi in forma matematica nell’era della proteomica, ha fornito lead, quindi, sono ligandi che tipica-
consente la scoperta di nuovi farmaci. decine di migliaia di strutture (serrature) mente presentano un’affinità di legame
L’uso di tecniche sperimentali comple- mediante tecniche cristallografiche e sub ottimale rispetto quella desiderata.
mentari all’informatica aumenta la microscopiche che, con l’introduzione Generalmente, un composto lead,
probabilità di successo in molte fasi del dei metodi NMR e bioinformatici rispetto il farmaco da questo generato,
processo di scoperta del farmaco, dall’i- permettono l’identificazione e l’analisi presenta una minor complessità struttu-
dentificazione di nuovi recettori, e delu- di siti attivi, proponendone eventuali rale, peso molecolare inferiore, minor
G Med Mil. 2013; 163(1): 31-48 33G
M
M
numero di anelli e legami ruotabili, infe- In figura 4 ed in figura 5 sono figura 6. Come sappiamo, la neuramini-
riore idrofobia (espressa come logP e riportati i metodi computazionali di dasi è un potenziale bersaglio contro il
LogD), infine hanno polarizzabilità infe- procedure CADD. virus dell’influenza in quanto rappresenta
riore. Infine, da un punto di vista un punto critico nel ciclo vitale del virus:
pratico, al fine di considerare l’ulteriore al temine della replicazione all’interno della
sviluppo del processo dell’ottimizza- Esempi storici cellula ospite, questo enzima permette ai
zione della biodisponibilità e sicurezza, virioni di fuoriuscire dalla cellula
i composti leads dovrebbero possedere Zanamivir e gli inibitori delle rompendo il legame tra l’emoagglutinina
le seguenti proprietà: proteasi HIV sono un paio di classi virale e l’acido sialico presente sulla super-
1 Caratteristiche chimiche relativa- esempi di CADD. ficie della cellula ospite. Venne poi
mente semplici (ottimizzazione Zanamivir. scoperto che il sito attivo dell’enzima è
chimica combinatoriale); Lo zanamivir (8) fu sviluppato nell’am- estremamente conservato in tutti i ceppi
2 Appartenenza ad una classe RSA bito di una collaborazione internazionale. virali, umani ed animali, mentre lo scaffold
(relazione struttura-attività) ben Nel 1983, la struttura tridimensionale presenta macroscopiche differenze strut-
conosciuta; dell’enzima neuraminidasi venne risolta turali; essendo già noto un inibitore sinte-
3 Situazione brevettuale favorevole; mediante cristallografia a raggi X, strutture tico della neuroaminidasi, analogo dell’a-
4 Buona ADME. pdb 1NNA ed 1NNB (9) rappresentate in cido sialico, ed essendo stata risolta la strut-
Genomica
Identificazione Validazione del Scoperta del Ottimizzazione
connessa alla Fase preclinica Fase clinica
del bersaglio bersaglio lead del lead
malattia
Bioinformatica; Druggability del Libreria di ligandi; QSAR; In silicio ADMET;
Reverse docking; recettore Docking; 3D - QSAR; Simulazioni
Predizione struttura De novo design; Structure based farmacocinetiche.
proteica Farmacoforo; drug design.
Induzione del
recettore.
Fig. 4 - Fasi della scoperta di nuovi farmaci e relazione delle tecniche informatiche strutturali.
Fig. 5 - Attività connesse con il drug design.
34 G Med Mil. 2013; 163(1): 31-48G
M
M
tura tridimensionale del suo complesso
con la neuraminidasi, i ricercatori riusci-
rono a disegnare in silico un lead che a
seguito di CADD permisero la realizza-
zione di un primo potenziale inibitore
guanidino simile che, anche se difficil-
mente sintetizzabile, aveva una cinetica di
complessazione molto favorevole (inibi-
zione a concentrazioni nano molari, sintesi
descritta in figura 7). Lo Zanamivir fu
brevettato dalla GlaxoSmithKline Inc. nel
1990 ed autorizzato al commercio dalla
FDA nel 1999. Successivamente, con meto-
diche analoghe, un’altra major farmaceu-
tica, Roche, brevettò l’oseltamivir (10), altro
importante inibitore della neuroaminidasi.
Inibitori della proteasi HIV (2).
In analogia con quanto detto per
zanamivir e oseltamivir, lo sviluppo
degli inibitori della proteasi HIV iniziò
durante la fine degli anni ’80. Ritonavir
(Norvir) è uno dei primi esempi in cui
venne applicata la genomica per il
Fig. 6 - Complesso di neuraminidasi ed acido 2-deossi 2,3-deidro-N-acetil neuramminico.
Immagine F. Spagnolo. drug design. Quando fu pubblicata la
Fig. 7 - Schema sintetico della sintesi dello zanamivir (1).
G Med Mil. 2013; 163(1): 31-48 35G
M
M
sequenza del genoma dell’HIV a metà Virtual screening consente di tener conto anche dell’in-
anni ’80, vennero identificate speci- duzione reciproca di ligando-recettore,
fiche sequenze nucleotidiche indica- Le procedure di virtual screening infatti, quasi tutti i programmi consen-
tive della codifica per enzimi protea- necessitano di un modello 3D del recet- tono di tener conto della flessibilità degli
sici. Kempf et. Al scoprirono successi- tore su cui si voglia disegnare un amminoacidi presenti nei siti attivi. Per
vamente che la proteasi è composta farmaco. I modelli 3D possono prove- trattare la flessibilità del ligando e del
da un dimero format da due metà nire da strutture risolte sperimental- sito attivo si sono sviluppate tre princi-
perfettamente identiche alla cui inter- mente (cristallografia raggi X, NMR, pali categorie di algoritmi: metodi siste-
faccia è allocato il sito attivo – detta- microscopia…) o virtuali (homology matici (sono considerate sistematica-
glio rilevante svelato in silico prima modelling). La filosofia del virtual scree- mente tutte le conformazioni del sistema
che la struttura tridimensionale fosse ning si basa sul test di affinità tra ligando in esame), stocastici (metodi Monte
risolta – con simmetria C2 (la rotazione e recettore su una libreria di molecole Carlo, algoritmi genetici) e simulazioni
di 180 gradi attorno all’asse centrale potenzialmente attive. Ogni ligando (dinamica molecolare e minimizzazione
porta ad una struttura identica). Con pertanto interagisce virtualmente con il dell’energia). Esistono decine di
questi dati, Kempf creò un modello recettore ed il computer determina un programmi per il docking (11, 12, 13,
computazionale del sito attivo protea- punteggio di questa interazione tenendo 14, 15, 16). I metodi per calcolare il
sico e disegnò potenziali inibitori in conto di parametri geometrici ed elet- legame ligando-recettore devono
silico. Partendo da un substrato noto trostatici. Nonostante si debba per forza tuttavia sottostare al compromesso di
fu generata facilmente una nuova fami- ricorrere a punteggi “approssimati”, al ottimizzare velocità ed accuratezza di
glia di composti dotati di una marcata giorno d’oggi è possibile stimare in calcolo mediante approssimazioni o stra-
attività inibente. L’uso congiunto delle maniera corrispondente alla realtà tagemmi informatici. Infatti, se ipotizzas-
strutture tridimensionali di proteasi sistemi complessi, anche grazie all’uso simo che la stima dell’affinità di legame
HIV man mano risolte, e della grafica, di modelli quantomeccanici. tra un ligando ed un recettore servisse
gli inibitori della HIV proteasi furono un giorno, testare un milione di
ottimizzati virtualmente anche in Procedure computazionali su recettori composti sullo stesso recettore richiede-
termini di biodisponibilità. Il primo sperimentalmente noti rebbe anni. Tra gli stratagemmi messi a
composto con sufficiente biodisponi- Il docking molecolare è il metodo punto, nel 2000, Arthur J. Olson, PhD,
bilità orale è il ritonavir (1991), appro- preferibile per lo studio dell’interazione professore di biologia molecolare e
vato dall’FDA a tempo di record (72 ligando-recettore quando la struttura di direttore del laboratorio di grafica mole-
giorni, anno 1996). Il tempo richiesto quest’ultimo è nota. Il docking è un algo- colare de “The Scripps Research Insti-
per lo sviluppo di questa classe di ritmo di calcolo che indica di come un tute”, avviò il progetto FightAids@Home
farmaci fu circa la metà di quello ligando interagisce con un recettore e che utilizza un sistema di calcolo a
richiesto per lo sviluppo con metodo- produce una stima dell’intensità di matrice via internet per effettuare Virtual
logie classiche – 8 anni –. Saquinavir legame. In un algoritmo di docking sono Screening da remoto su personal
e nelfinavir sono altri inibitori realiz- perseguiti due obiettivi principali, il computers di volontari. Il progetto può
zati negli stessi anni. Gli inibitori delle primo, la predizione delle pose del essere riassunto in questi termini:
proteasi HIV sono un passo miliare ligando (conformazione della struttura 500,000 volontari installarono una specie
nella rivoluzione del drug design e e topologia di interazione), il secondo, di salva schermo che si attivava, e
della terapia anti HIV. la corretta classifica di interazione lanciava calcoli del programma Auto
Dallo Zanamivir i metodi utilizzati ligando-recettore. Mentre il primo obiet- Dock, nei momenti di standby del pc,
per il drug design si sono evoluti fonda- tivo ha da tempo trovato soluzioni in sei mesi, degli oltre 2,000 ligandi che
mentalmente in due direzioni: Virtual robuste, il secondo presenta dei limiti vennero testati su centinaia di strutture
Screening la prima, fragment based connessi con la tipologia di approssima- proteiche di HIV, alcune decine sono in
design la seconda. Di seguito vengono zioni adottate nella ricerca della solu- fase avanzata di studio. Oltre all’enorme
descritte le filosofie alla base delle due zione ottimale per la descrizione del risparmio di risorse computazionali,
metodologie. sistema specifico. Inoltre, il docking questo progetto fu pionieristico per
36 G Med Mil. 2013; 163(1): 31-48G
M
M
quanto concerne i dati statistici raccolti ligando, in seconda battuta sono alli- degli approcci di modellazione QSAR
che sono serviti per identificare sottoca- neati i ligandi nelle varie conformazioni è l’identificazione una nuova classe di
tegorie di proteine mutanti, infatti fu per definire quali caratteristiche inibitori dell’iston-deacetilasi tra 9,5
possibile definire le caratteristiche chimiche sono comuni ed essenziali. milioni molecole presenti nei database
comuni che devono possedere non solo Esistono due tipi di modelli farmacofo- ZINC, World Drug Index, le librerie
gli inibitori retro virali, ma anche inibi- rici: nel primo tipo la relazione strut- Synergy ASINEX, e altre banche dati
tori del recettore delle citochine (antitu- tura attività quantitativa tridimensionale commerciali (17, 18, 19, 20).
morali) oppure antibiotici diretti al ribo- (QSAR-3D) tiene conto dei dati speri-
soma. I composti candidate per i test in mentali di relazione struttura-attività di
laboratorio sono passati in esame da un un set molto vasto di molecole (non Homology modeling
operatore che li seleziona e li studia ulte- necessariamente di dimostrata attività Quando non è proprio disponibile
riormente mediante simulazioni dina- biologica), nel secondo tipo sono consi- una struttura risolta del recettore su
miche del complesso ligando/recettore. derati una selezione di principi attivi. cui sviluppare il farmaco, ma è nota
Con i modelli farmacoforici è possibile la sequenza amminoacidica – o
Procedure computazionali su recettori determinare qualitativamente le nucleotidica del gene che la codifica
virtuali potenze dei composti testati. I punti – la modellazione per omologia (noto
QSAR determinanti per il modelling farma- anche come modellazione compara-
Nel caso in cui la struttura recetto- coforico sono la definizione e posizio- tiva) è un approccio utile per spiegare
riale non fosse nota, i chimici compu- namento dei descrittori farmacoforici e dati sperimentali, sviluppare ipotesi
tazionali utilizzano metodi definiti le tecniche di allineamento utilizzate oppure effettuare Structure-Based
ligand-based: sovrappongono una serie dall’algoritmo. I modelli QSAR sono più Drug Design (SBDD). Nella modella-
di ligandi con attività nota contro il performanti in termini di tempo di zione per omologia viene costruito un
bersaglio e confrontano le loro caratte- esecuzione del calcolo, pertanto sono modello a risoluzione atomica della
ristiche strutturali e chimiche. Uno dei preferibili quando il problema che proteina bersaglio partendo da
modelli più comuni per il ligand-based viene posto al ricercatore è rappresen- sequenze amminoacidica sperimentali
è il farmacoforico dove gruppi funzio- tato da un database molto grande. I note correlate omologhe che vengono
nali caratteristici (e.g. donatori di risultati ottenuti generalmente consi- utilizzate come stampo (21, 22, 23). Il
legame idrogeno, cariche elettrosta- stono in una clusterizzazione di poten- concetto si basa sull’esperienza che
tiche…) sono usati per generare una ziali ligandi nuovi e di diversa tipologia sequenze simili portano a strutture
impronta per selezionare virtualmente chimica, ciò consente al chimico farma- simili e verosimilmente discendono da
altri composti simili. In questo caso, ci ceutico di includere un nuovo spazio un antenato comune (una famiglia di
si riferisce ad un modello chiave-serra- chimico nella fase di ricerca. Un proteine), pertanto hanno sequenze
tura adattato alla ricerca di una vera e modello farmacoforico tridimensionale amminoacidiche e strutture 3D simili.
propria impronta tridimensionale e può essere proposto anche conside- Poiché la determinazione sperimentale
pseudo complementare al sito di inte- rando la struttura del recettore preso in della struttura delle proteine è ancora
razione con il recettore. Infatti, le super- considerazione. In questo caso viene un processo difficile e costoso, metodi
fici di ligando e del recettore si compor- studiata la forma ed il volume del recet- di modellazione di omologia forni-
tano come il calco ed il suo stampo, tore considerandolo “stampo” in nega- scono modelli adeguatamente affida-
con in più l’interazione elettrostatica. tivo per lo sviluppo del rispettivo bili rapidamente. Tipicamente, la
Ne consegue che un principio attivo ligando. Il 3D QSAR consente pertanto modellazione per omologia delle
potrebbe legare diversi recettori anche di effettuare una prima selezione tra proteine comprende le seguenti
dotati di attività diverse tra loro. La milioni di composti di cui sono più quattro fasi (21, 22, 23): identificazione
generazione di un modello farmacofo- facilmente predicibili le proprietà di una o più note strutture sperimen-
rico ligand-based avviene in due stadi, chimico-fisiche, il che consente di tali di una proteina correlata che può
per prima cosa viene campionato lo proporre un profilo ADMET. Un servire come modello, allineamento
spazio conformazionale di ogni esempio pratico sulla significatività delle sequenze di proteine bersaglio e
G Med Mil. 2013; 163(1): 31-48 37G
M
M
stampo, costruzione del modello per Fragment based drug design glivec. Lo screening dei frammenti della
il target ed infine la raffinazione / vali- loro libreria contro questa struttura
dazione / valutazione dei modelli otte- La filosofia di questo approccio di permise l’individuazione di un primo
nuti. L’intervento umano è in genere drug design si ispira ai famosi matton- lead attivo già a concentrazioni micro
necessario per verificare la presenza cini “Lego”. In un laboratorio, è normale molare e, dopo soli tre mesi, grazie alle
di errori che possono essere stati intro- che i ricercatori creino le proprie librerie successive modifiche suggerite dall’ana-
dotti nel corso, ad esempio, l’allinea- di composti o frammenti che rappresen- lisi computazionale, fu isolato un
mento di sequenza o la robustezza dei tano unità funzionali facilmente assem- composto in grado di inibire la BCR-AB
modelli. FASTA (24) e BLAST (25) sono blabili tra loro. Per ogni composto della a concentrazioni nano molari.
i metodi di ricerca in database più libreria sono raccolti dati sperimentali
semplici per individuare i modelli per ottenuti secondo varie metodologie
la modellazione omologia. Strumenti (cristallografia ad alta efficienza, NMR o Obiettivi “Complicati”
più avanzati includono PSI-BLAST (26) spettrofotometria di massa) per descri-
e FFAs (27). La qualità di un modello vere la conformazione 3D della struttura Lo studio dell’interazione ligando-
ottenuto per omologia è generalmente considerata. Successivamente, con l’au- recettore tout-courts, per quanto inda-
correlata con quella della struttura di silio di tecniche computazionali, i fram- ginoso possa essere, valuta due “oggetti”
riferimento e la procedura di allinea- menti sono trasformati in potenti farmaci finiti e di facile individuazione. Quando
mento. Quando sono presenti lacune a seguito dell’aggiunta di gruppi funzio- questi però sono inseriti in un ambiente
(indels) tra struttura di riferimento e nali o piccole modificazioni. Questo biologico più completo, quale può
modello, la modellazione per approccio trae il massimo rendimento essere il doppio strato fosfolipidico delle
omologia può essere soggetta a errori, con l’accoppiamento alle tecniche della membrane cellulari, il grado di comples-
inoltre, la qualità del modello, tende chimica combinatoriale. Il vantaggio più sità aumenta enormemente in quanto
a diminuire se la risoluzione della evidente sta nella semplificazione del non si può considerare solo il sito attivo
proteina modello è scarsa, tuttavia, dal sistema complesso che permette l’esplo- del recettore, ma bisogna simulare anche
momento che la regione funzionale razione sistematica dello spazio chimico eventuali interazioni non solo con la
della proteina, come il sito attivo, è con un numero limitato di frammenti membrana stessa, ma anche con altre
altamente conservata, i modelli otte- (e.g. l’esplorazione di 10 frammenti nelle proteine. Oltre alla difficoltà computa-
nuti per omologia sono significativa- tre dimensioni x, y, z, permette di esplo- zionale del problema, si aggiunge la
mente rappresentativi del recettore rare 1,000 possibili combinazioni). Uno complicazione sperimentale della riso-
studiato (28). Un modello omologo dei pionieri dei metodi Fragment Based luzione delle strutture recettoriali. Un
ragionevole può essere quantificato, è Sir. Tom Blundell della cattedra di dato significativo della sfida posta alla
per esempio, da una trama Ramachan- biochimica dell’Università di Cambridge. ricerca è rappresentato dalla propor-
dran, in cui viene mostrata la distribu- Nel 1999 avviò uno studio sistematico zione dei farmaci sul mercato che ha
zione di angoli di legame del back- per la valutazione di nuovi inibitori di come bersaglio recettori accoppiati a
bone proteico e dalla distribuzione dei kinasi a scopo antitumorale. Il connubio proteine G: 1 su 4!
residui amminoacidici idrofili e lipo- tra la cristallografia a raggi X ad alta effi-
fili. Per valutare la qualità relativa dei cienza e tecniche computazionali,
modello omologi esistono numerosi permisero la scoperta di un altro Trasformazione di composti
esperimenti di benchmarking su larga farmaco innovativo: il glivec (33) un chimici in farmaci
scala della predizione di strutture inibitore del BCR-ABL, chinasi perenne-
proteiche (CASP) (29). Un modello mente attivato e coinvolto nella leucemia I composti che sono stati identificati
ottenuto per omologia può essere cronica mielogena. Dal momento che con metodi computazionali come possi-
utilizzato per la valutazione di drug- circa il 20% dei pazienti sono resistenti bili principi attivi sono ben lungi dall’es-
gability ed esperimenti di mutagenesi, al farmaco, i ricercatori della SGX clona- sere considerati farmaci. Per essere un
ma deve essere applicato con cautela rono, espressero, purificarono e cristal- farmaco un composto chimico deve
per sviluppo di farmaci (30, 31, 32). lizzarono una proteina resistente al essere biodisponibile e sicuro. Proprio
38 G Med Mil. 2013; 163(1): 31-48G
M
M
queste due caratteristiche portano a scientifica, inclusa l’industria. Questi atomi e molecole come sfere e molle,
derive nella formulazione della struttura database possono raccogliere insiemi di governati dalle leggi della meccanica
chimica finale e, pertanto, molto si è fatto strutture, come ad esempio dei campioni classica e non come nuclei tenuti
per valutare in silico la biodisponibilità di screening disponibili in commercio, insieme da orbitali elettronici gover-
e disponibilità. I modelli matematici per oppure fornire dati aggiuntivi come la nati dalle leggi della meccanica quan-
la predizione di questi requisiti sono bioattività dei composti e dei loro target tistica, come sono realmente. Attual-
ormai abbastanza maturi e possono proteici. In Tabella 6 è riportata una sele- mente, con l’aumento delle prestazioni
simulare con una buona approssima- zione di alcuni dei più noti database di dei calcolatori ed il miglioramento di
zione gli effetti biologici di un composto composti utilizzati per la scoperta di algoritmi e software, i problemi biolo-
nel nostro organismo. Rispetto la famosa nuovi farmaci. Il mantenimento di gici possono essere affrontati con
regola dei 5 che ha formulato Lipinsky queste raccolte è possibile grazie alla metodi quantomeccanici (36, 37, 38).
per descrivere le caratteristiche chimico- creazione di software chemoinformatica I metodi quantomeccanici possono
fisiche minime di un farmaco, i computer utilizzare la correttezza delle informa- essere utilizzati per modellare mole-
sono oggi in grado di fornire indicatori zioni riportate (34). cole instabili, come i radicali, o per
sufficientemente rigorosi per determinare fornire stime di energie di attivazione
l’ADME in silico, ossia la dose assorbita Dinamica molecolare per le reazioni chimiche, comprese
dal corpo, la distribuzione negli organi, La dinamica molecolare è molto utile quelle enzimatiche. Le applicazioni
il metabolismo e l’escrezione di una per comprendere il comportamento quantomeccaniche tipiche nella
molecola. Con sufficiente potenza di delle proteine o altre macromolecole progettazione di farmaci includono:
calcolo, gli scienziati possono anche biologiche. Grazie all’enorme aumento l’ottimizzare di ligandi e complessi
testare virtualmente un composto candi- di potenza di calcolo e algoritmi miglio- recettore-ligando (39), costruzione di
dato a diventare farmaco su tutto l’in- rati, con i metodi di dinamica moleco- modelli QSAR.
sieme delle strutture risolte e, mediante lare si possono simulare le dinamiche
metodi di homology modelling, anche su di sistemi complessi di grande dimen- Programmi per il calcolo di ADMET
recettori virtuali, per essere avere l’indi- sioni anche in ambienti di solvente espli- I programmi per li calcolo di
cazione che il lead attraversi le giuste cito (35). Nonostante l’ampio uso di ADME sono molto importanti nello
membrane, non reagisca con recettori simulazioni di dinamica molecolare sviluppo di un nuovo farmaco al fine
indesiderati e che abbia l’attività voluta nello sviluppo di farmaci, la messa a di ridurre il fallimento in fase avan-
sul proprio bersaglio. In questi termini punto di una simulazione di dinamica zata e minimizzare i costi (40) (41).
potrebbe sembrare che il computer abbia molecolare può essere difficile: spesso Numerose proprietà ADMET sono
ormai sostituito radicalmente il banco di non esistono parametri adeguati per il interdipendenti fra loro, quindi è
laboratorio. Il chimico farmaceutico calcolo, come ad esempio nel caso dei necessario ottimizzarle contempora-
continua ad essere indispensabile per il composti metallorganici. Infine, visto il neamente durante lo sviluppo di un
valore aggiunto derivante dall’intuizione, tempo richiesto dalle simulazioni, non farmaco. L’ottimizzazione ADMET
o senso chimico, che lo contraddistingue è possibile effettuare dei calcoli siste- multiparametrica è, probabilmente, la
da un calcolatore. matici su grossi database, pertanto, con fase meno attraente ma può fare la
la dinamica molecolare è possibile rifi- differenza costosa tra successo e falli-
nire i risultati già ottenuti tramite le altre mento. Molti programmi usano
Strumenti virtuali per procedure metodologie sopra descritte. modelli statistici per calcolare i
CADD descrittori ADMET. La qualità dei
Programmi di meccanica quantistica modelli dipende molto dalla giusta
Database chimici Gli approcci già descritti sono combinazione di tecniche statistiche,
La quantità di informazioni generate caratterizzati dalle approssimazioni descrittori molecolari, metodo di vali-
dalla “meta-omica” ha reso necessaria la necessarie per la trattazione dei sistemi dazione, e soprattutto per la qualità e
standardizzazione e razionalizzazione secondo la meccanica molecolare, la l’ampiezza dei dati sperimentali utiliz-
dei dati in possesso dalla comunità più importante, è la descrizione di zati per derivarli (42).
G Med Mil. 2013; 163(1): 31-48 39G
M
M
8 V. G. R. P. S. C. F. D. F. L. F. S.
Programmi di visualizzazione di ciò la serie di farmaci emergenti per Gehlhaar DK:
molecolare l’epatite C. Il virus dell’epatite C fu Molecular recognition of the inhibitor
Ligandi e recettori possono essere scoperto nel 1989 ed è caratterizzato da AG-1343 by HIV-1 protease:
conformationally flexible docking by
visualizzati ed analizzati mediante una difficile coltivazione, inoltre, le infor-
evolutionary programming.
programmi di modellistica più o meno mazioni strutturali dei bersagli moleco- Chem Biol, vol. 2, n. 5, pp. 17-24, 1995.
complessi. generare grafica per pubbli- lari (polimerasi e proteasi HCV) vennero
cazioni o relazioni. Per la generazione risolte molto più tardi (metà anni ’90). 9 P. C. M. B. Y. S. C. L. W. A. G. Bossart-
Whitaker:
di immagini di alta qualità o anche Grazie alle tecniche computazionali, in
Three-dimensional structure of influenza
animazioni per presentazioni e pubbli- breve tempo la RNA polimerasi (43) è A N9 neuraminidase and its complex
cazioni, i cinque programmi ampia- stata individuata come il bersaglio ideale with the inhibitor 2-deoxy 2,3-dehydro-
mente utilizzati sono Chimera, Jmol, per lo sviluppo di nuovi inibitori, al punto N-acetyl neuraminic acid.
J.Mol.Biol., vol. 232, pp. 1069-1083, 1993.
PyMOL, SwissPdbViewer e VMD sono di diventare essa stessa un modello per
inclusi nella. L’ uso di questi software testare gli algoritmi ed architetture infor- 10 M. Karpf e R. Trussardi:
non è limitato alla visualizzazione mole- matiche più all’avanguardia. New, Azide-Free Transformation of
Epoxides into 1,2-Diamino Compounds:
colare in quanto sono altamente esten-
Synthesis of the Anti-Influenza
sibili per l’analisi di strutture molecolari Neuraminidase Inhibitor Oseltamivir
e dati correlati, tra cui mappe di densità Bibliografia Phosphate (Tamiflu).
elettroniche, assemblaggi supramoleco- Journal of Organic Chemistry, vol. 66, n.
1 A. I. T. S. a. S. K. i. C. H. a. T. F. H. 6, p. 2044–2051, 02 4 2001.
lari, allineamenti di sequenza, analisi di
Chuman:
docking e dinamiche molecolari. Classical and Three-Dimensional QSAR 11 G. D. H. R. Morris GM.:
in Agrochemistry. Automated docking using a Lamarckian
ACS Symposium Series 606, p. 171, 1995. genetic algorithm and an empirical
binding free energy function.
Conclusioni J Comput Chem, vol. 19, n. 14, p. 1639–
2 Scheigetz J., Zamboni R., Bernstein
M. A. e Roy B.: 1662, 1998.
La produttività dell’industria farma- A syntheses of 4-a-guanidino-2-deoxy-
2,3-didehydro n-acetylneuraminic acid. 12 B. S. M. S. Lang PT.:
ceutica è diminuita moltissimo negli
Organic Letters , vol. 27, n. 6, p. DOCK 6: combining techniques to model
ultimi dieci anni. Anche se questo è 637–644, 12 1995. RNA-small molecule complexes.
probabilmente dovuto a molti fattori, in RNA, vol. 15, n. 6, p. 1219–1230, 2009.
particolare, gli standard di sicurezza e 3 Online. Available:
http://fightaidsathome.scripps.edu/ 13 R. M. L. T. Kramer B.:
l’enorme costo e tempo delle sperimen-
fightaidsathome.scripps.edu. Evaluation of the FLEXX incremental
tazioni cliniche. La domanda evidente The Scripps Institute. construction algorithm for protein–ligand
che ci si può porre è legata al vero contri- docking.
4 http://www.worldcommunitygrid. Proteins, vol. 37, n. 2, p. 228–241, 1999.
buto del computational drug design, visto
org/research/gfam/overview.do
che i suoi costi, in ogni caso, non sono 14 A. H. N. A. G. J. B. F. Mcgann MR.
irrilevanti e, per di più non esiste un 5 P. L.: Gaussian docking functions.
programma che sia affidabile per tutti i Il ruolo di AIFA nella Ricerca Clinica. Biopolymers, vol. 68, n. 1, p. 76–90,
2012. 2003.
sistemi studiati (l’utilizzo di metodologie
di docking è stato vincente per la proget- 6 F. E.: 15 B. J. M. R. Friesner RA.:
tazione degli inibitori delle proteasi HIV Einfluss der Configuration auf die Glide: a new approach for rapid,
Wirkung der Enzyme. accurate docking and scoring. 1 Method
ma deludente in altri ambiti). È certo che
Ber. Dt. Chem. Ges., vol. 27, pp. 2985- and assessment of docking accuracy.
l’utilizzo del Computer Aided Drug 2993, 1894. J Med Chem, vol. 47, n. 7, p. 1739–1749,
Design ha avuto un grandissimo impatto 2004.
7 K. D. E.:
nella chimica farmaceutica specie per la
Application of a Theory of Enzyme 16 C. J. H. M. M. C. T. R. Verdonk ML.:
possibilità di virtualizzare e, a volte Specificity to Protein Synthesis.
Improved protein–ligand docking using
predire, fenomeni biologici difficilmente Proc. Natl. Acad. Sci., vol. 44, n. 2, pp.
GOLD.
riproducibili in laboratorio. Un esempio 98-104, 1958.
Proteins, vol. 52, n. 4, p. 609–623, 2003.
40 G Med Mil. 2013; 163(1): 31-48G
M
M
17 P. R. S. J. L. M. Sprous DG.: 26 Online. Available: 36 R. K. M. K. J. Peters MB.:
QSAR in the pharmaceutical research www.ebi.ac.uk/Tools/sss/psiblast. Quantum mechanics in structure-based
setting: QSAR models for broad, large European Bioinformatics Institute. drug design.
problems. Curr Opin Drug Discov Devel, vol. 9, n.
Curr Top Med Chem, vol. 10, n. 6, p. 27 Online. Available: 3, p. 370–379, 2006.
619–637, 2010. http://ffas.ljcrf.edu/ffas-
cgi/cgi/ffas.pl.
37 P. M. W. B. Raha K.:
18 C. RD.:
The role of quantum mechanics in
Prospective ligand- and target-based 3D 28 S. A. Baker D.
QSAR: state of the art 2008. structure-based drug design.
Protein structure prediction and
Curr Top Med Chem, vol. 9, n. 9, p. Drug Discov Today, vol. 12, n. 17-18, p.
structural genomics.
791–810, 2009. 725–731, 2007.
Science, vol. 294, n. 5540, p. 93–96,
2001.
19 G. A. Tropsha A.: 38 C. P. R. M. Cavalli A.:
Predictive QSAR modeling workflow, 29 P. J. J. R. F. K. Moult J. Target-related applications of first
model applicability domains, and virtual A large-scale experiment to assess protein principles quantum chemical methods in
screening. structure prediction methods. drug design.
Curr Pharm Des, vol. 13, n. 34, p. Proteins, vol. 23, n. 3, p. ii–v, 1995. Chem Rev, vol. 106, n. 9, p. 3497–3519,
3494–3504, 2007. 2006.
30 P. S. Cavasotto CN.
20 W. X. H. X. Tang H.:
Homology modeling in drug discovery: 39 N. M. Liao C.:
Novel inhibitors of human histone
current trends and applications. Tautomerism and magnesium chelation
deacetylase (HDAC) identified by QSAR
Drug Discov Today, vol. 14, n. 13, p. of HIV-1 integrase inhibitors: a
modeling of known inhibitors, virtual
676–683, 2009. theoretical study.
screening, and experimental validation.
J Chem Inf Model., vol. 49, n. 2, p. ChemMedChem, vol. 5, n. 7, p.
31 G. M. F. M. Kairys V.:
461–476, 2009. 1053–1066, 2010.
Using protein homology models for
structure-based studies: approaches to
21 P. S. Cavasotto CN.: 40 H. T. R. A. Wang J.:
model refinement.
Homology modeling in drug discovery: Annu Rep Comput Chem.
Scientific World Journal, vol. 6, p.
current trends and applications. Chapter 5 Recent advances on in silico
1542–1554, 2006.
Drug Discov Today, vol. 14, n. 13-14, p. ADME modeling, vol. 5, 2009, p.
676–683, 2009.
32 S. T. Kopp J.: 101–127.
Automated protein structure homology
22 G. M. F. M. Kairys V.:
modeling: a progress report. 41 H. A. H. S. Gleeson MP.:
Using protein homology models for
structure-based studies: approaches to Pharmacogenomics, vol. 5, n. 4, p. In-silico ADME models: a general
model refinement. 405–416, 2004. assessment of their utility in drug
ScientificWorldJournal, vol. 6, p. discovery applications.
1542–1554, 2006. 33 «http://www.innovation.org/ Curr Top Med Chem, vol. 11, n. 4, p.
index.cfm/StoriesofInnovation/Innov 358–381, 2011.
23 S. T. Kopp J. atorStories/The_Story_of_Gleevec,»
Automated protein structure homology Online. 42 E. WJ.:
modeling: a progress report. Chapter 29.Computational models for
Pharmacogenomics, vol. 5, n. 4, p. 34 I. W. N. M. Sitzmann M.:
ADME.
405–416, 2004. Tautomerism in large databases.
Annu Rep Med Chem, 2007, p. 449–467.
J Comput Aided Mol Des, vol. 24, n. 6-7,
24 Online. Available: p. 521–551, 2010.
43 I. V. F. J. B. N. K.-B. R. K. M. C. N. I. F.
http://fasta.bioch.virginia.edu.
35 L.-L. K. D. R. S. D. Klepeis JL.: T. Iwona E. Weidlich.:
University of Virginia.
Long-timescale molecular dynamics Inhibitors for the hepatitis C virus RNA
25 «Online. Available: simulations of protein structure and polymerase explored by SAR with
http://blast.ncbi.nlm.nih.gov. function. advanced machine learning methods.
The National Center for Biotechnology Curr Opin Struct Biol, vol. 19, n. 2, p. Bioorganic & Medicinal Chemistry, vol.
Information. 120–127, 2009. 21, n. 11, pp. 3127-3137, 1 06 2013.
G Med Mil. 2013; 163(1): 31-48 41G
M
M
In silico techniques for drug design
Ferdinando Spagnolo *
Introduction inhibiting the dimer. Picture F. The rational drug design
Spagnolo.); in 1997 a derivative of the
The first computers were built in the second generation, Nelfinavir (1997) (2), The discovery and development of
40s for the description of atomic and was designed using docking, one of the new drugs is an intense interdisciplinary
molecular systems in the field of the most futuristic computational techni- effort. Generally, the discovery of new
military. In the last two decades of the ques of the time. By now, in the drugs is depicted as a linear process, in
twentieth century computational tech- scenario of medicinal chemistry, there succession, beginning with the discovery
niques have reached full maturity in the is no example of drug, developed or of the biological target and the lead
application of chemical systems, under development, which has not been compound (molecule with a specific
physical, biological and chemical-phar- influenced by at least one computa- coarse biological activity and which can
maceutical. In recent years Information tional methodology. The cost rationali- be further modified in order to modulate
Technology (IT) supported the tradi- zation and optimization of the process the biological effects). The optimization
tional techniques used in the laboratory of development that characterize the of the lead proceeds until the pre-clinical
and in the design of new drugs, both chemical-pharmaceutical and computa- phase, in vitro and in vivo, to determine
in the discovery or optimization of tional techniques, certainly offer whether the compounds elaborated meet
existing ones. Among the successes of valuable contributions to the develop- a series of pre-set criteria for the initia-
drugs designed with the aid of in silico ment of orphan drugs or those drugs tion of clinical development. The need
techniques, we can cite the Norfloxacin useful for the treatment of diseases that for optimization of resources for the deve-
(1), forefather of the fluoroquinolones, are very rare, given the low impact lopment of a new drug is implicit when
designed in the 80s, one of the most statement, are not central to the inte- analyzing the cost, time, and the demand
important families of antibacterials that rests of research and development in for curative drug for a specific disease. In
sees, among others, Ciprofloxacin one the pharmaceutical industry. The perfor- fact, the number of years to bring a drug
of the most important derivatives. Few mance of modern computers and acces- from discovery to market takes about 12-
years later, in 1994, was designed to sibility of computing resources have 14 years and have an approximate cost
Indinavir, inhibitor of a key enzyme for allowed, among other things, to start of between $ 1.2 and $ 1.4 billion. (5)
the vitality of the acquired immunode- non-profit projects on an international dollars (Fig. 3 - Phases of the development
ficiency virus type I (HIV-I), represented scale for the development of new drugs of a new drug compounds. Out of 5,000
with its receptor, HIV-1 polymerase, against malaria, tuberculosis, leishma- drug candidates, on average, only one is
(Fig. 2 - 3D visualization of the struc- niasis and AIDS (eg. fight AIDS project released in the market. The usefulness of
tures HSG1.pdb and indinavir. HSG1 is @ home (3) and GO Fight Against the drug design can already be seen in
the three-dimensional model of HIV-1 Malaria (4) ...). The main limitations of the preliminary study of lead candidate
polymerase. In the figure, the dimer is computational aided drug design compounds so that only a fraction of the
shown in blue cartoon, chain A, and (CADD) reside in the inherent difficulty hundreds of thousands of compounds are
green, chain B. The active site of the of developing mathematical models to proposed to be short, then continues giving
enzyme is placed at the interface of the represent sufficiently complete in its analytical tools for the analysis of the
dimer. In this model, the indinavir is entirety complex biological systems at results of preclinical testing and, nowa-
positioned in the active site irreversibly the base of cell physiology. days also trials).
* Cpt. (pharm.) Army Health and Veterinary Center of Study and Research, Roma.
PhD. in Biomedical and Biotechnology – Bioinformatics – University of Udine.
42 G Med Mil. 2013; 163(1): 31-48G
M
M
Traditionally, drugs were discovered 1 Virtual Screening and de novo safety, the lead compounds should
by synthesizing compounds in multi- design; possess the following properties:
step processes where the active 2 In silico ADME / T prevision; 1 Chemical characterization relatively
compounds were tested on selected 3 Characterization of receptor / ligand simple (optimization by means of
biological systems, first in vitro, then in interaction. combinatorial chemistry);
vivo, to further select the most promi- With this in mind, one of the key 2 Well known RSA class;
sing lead candidates for their pharma- principles for the modern computational 3 Favorable patent situation;
cokinetic properties, metabolism and drug design, remains, in any case, the Good ADME.
potential toxicity. Such a development key-lock model (6), then developed as Figure 4 (Stages of drug discovery
process is characterized by high rates of a model of adaptation induced (7). and structural relationship of informa-
failures attributable to poor pharmacoki- Molecular biology, in the era of proteo- tion technology) and Figure 5 (Attività
netics, lack of efficacy, animal toxicity, mics, has provided tens of thousands of connesse con il drug design) show the
side effects in humans and various structures (locks) by crystallographic methods of computational procedures
commercial factors. Today, the process and microscopic techniques that, with CADD.
of drug discovery has been revolutio- the introduction of the methods NMR
nized with the advent of genomics, and bioinformatics tools allowed the
proteomics, bioinformatics and high-effi- identification and analysis of active sites, Historical examples
ciency technologies such as, combina- proposing any conformers, suggesting
torial chemistry, high throughput scree- potential pharmacologically active Zanamivir and HIV protease inhibi-
ning (HTS), virtual screening, de novo ligands (keys). The resolved structure of tors are a couple of examples of CADD
design, ADMET (Absorption, Distribu- a ligand-receptor complex provides a classes.
tion, Metabolism, Elimination, Transfor- detailed view of the interactions Zanamivir
mation) in vitro and in silico screening. between the ligand and the receptor. Zanamivir (8) was developed as
Virtual methods can help to identify the The detailed study of a compound with part of an international collaboration.
targets of the active principle over that drug-like characteristics allows to In 1983, the three dimensional struc-
describe the physical-chemical proper- propose structural changes are able to ture of the enzyme neuraminidase was
ties. The tools common to all computa- modulate the formation of the ligand- resolved by X-ray crystallography, pdb
tional methods are management systems receptor complex consists of modifying structures 1NNA and 1NNB (9) repre-
(database optimized), high-performance the ADME. sented in figure 6 (Neuraminidase
scientific computing (High Performance A drug-like compound (or drug- acid 2-deoxy 2,3-dehydro-N-acetyl
Computing - HPC) and the Internet. gable) contains functional groups and neuraminic acid. complex. Image F.
Access to an unimaginable amount of / or possesses chemical and physical Spagnolo). As we know, the neurami-
information and the processing of properties in line with most of the nidase is a potential target against
complex biological data in a mathema- known drugs. Lead structures, influenza virus where it represents a
tical form allows the discovery of new therefore, are ligands that typically have critical point in the life cycle of the
drugs. The use of complementary expe- a binding affinity than the desired sub- virus. During the replication within the
rimental techniques to information optimal. Generally a lead compound, host cell, this enzyme allows virions to
increases the likelihood of success in than the drug generated by this, escape from the cell by breaking the
many stages of the drug discovery presents a low structural complexity, bond between the viral hemagglutinin
process, from the identification of new low molecular weight, few rings and and sialic acid on the surface of the
receptors, and elucidation regarding rotatable bonds, low hydrophobicity host cell. It was later discovered that
their functions, to the discovery and (expressed as logP and logd), finally the active site of the enzyme is highly
development of leads with the desired has low polarizability. From a practical conserved in all strains, human and
properties. The main roles of scientific point of view, in order to consider the animal, while the scaffold presents
computing in the discovery of an active further development of the process of macroscopic structural differences.
compound are: optimizing the bioavailability and Since it was already known a synthetic
G Med Mil. 2013; 163(1): 31-48 43G
M
M
inhibitor of neuraminidase, an HIV protease gradually resolved, and Computational procedures of experi-
analogue of sialic acid, and the three- graphics, inhibitors of HIV protease mentally known receptors
dimensional structure of its complex were virtually optimized eventually in Molecular docking is the preferable
with the neuraminidase was resolved, terms of bioavailability. The first method to study ligand-receptor inte-
the researchers were able to draw in compound with adequate oral bioavai- raction when the structure of the latter
silico a lead that following CADD lability is ritonavir (1991), approved by is known. Docking is a calculation algo-
allowed the realization of a first poten- the FDA in record time (72 days, year rithm that indicates how a ligand inte-
tial inhibitor guanidino similar that, 1996). The time required for the deve- racts with a receptor and produces an
although hardly synthesized, it had a lopment of this class of drugs was estimate of the intensity of binding. In
very favorable kinetics of complexa- approximately half of that required for a docking algorithm are pursued two
tion (inhibition to nano molar concen- development with classical methodo- main objectives, the first, the predic-
trations, the synthesis described in logies - 8 years -. Other inhibitors tion of the poses of the ligand (confor-
figure 7 - Zanamivir sinthetic path saquinavir and nelfinavir are made in mation of the structure and topology
(1)). The Zanamivir was patented in the same years. The HIV protease inhi- of interaction), the second, a classifica-
1990 by GlaxoSmithKline Inc. and bitors are a landmark step in the revo- tion of ligand-receptor interaction.
licensed to trade by the FDA in 1999. lution of drug design and anti-HIV While the first objective has robust solu-
Then, by similar methods, another therapy. tions, the second has limitations asso-
major pharmaceutical Roche, patented From zanamivir the methods used ciated with the type of approximations
oseltamivir (10), another important for drug design have evolved in two made in the search of the optimal solu-
neuraminidase inhibitor. basic directions: Virtual Screening the tion for the description of the specific
HIV protease inhibitors (2) first, fragment based design the second. system. In addition, docking allows to
In analogy with what it was said The philosophies behind the two metho- also take into account mutual induction
for zanamivir and oseltamivir, the deve- dologies are below presented. of ligand-receptor interaction, in fact,
lopment of HIV protease inhibitors almost all of the programs allow you
began during the late ‘80s. Ritonavir Virtual screening to take into account the flexibility of
(Norvir) is one of the first examples in the amino acids present in the active
which it was applied genomics for drug The procedures of virtual screening sites. To treat the flexibility of the ligand
design. When it was published the require a 3D model of the receptor on and the active site three major classes
genome sequence of HIV in the mid which design a drug. The 3D models of algorithms were developed: syste-
80s, were identified indicative of can be structures solved experimentally matic methods (are systematically
specific nucleotide sequences coding (X-ray crystallography, NMR, micro- considered all the conformations of the
for enzymes protease. Kempf et. Al scopy...) or virtual (homology mode- system under consideration), stochastic
subsequently discovered that the ling). The philosophy of the virtual (Monte Carlo methods, genetic algo-
protease is a dimer composed of two screening is based on the affinity rithms) and simulations (molecular
monomers perfectly identical holding between ligand and receptor on a dynamics and minimization energy).
at the interface the active site - rele- library of potentially active molecules. There are dozens of programs for
vant detail revealed in silico before the Therefore each ligand interacts with the docking (11, 12, 13, 14, 15, 16). The
three-dimensional structure was solved receptor and virtually the computer methods to calculate the ligand-
- with C2. With these data, Kempf determines a score for this interaction receptor are, however, subject to
created a computational model of the taking into account the geometric para- compromise to optimize speed and
active site and drew potential protease meters and electrostatic. Although it accuracy of calculation by computing
inhibitors in silico. Starting from a must necessarily resort to scores approximations or gimmicks. In fact, if
known substrate was easily generated “approximate”, nowadays it is possible we assumed that the estimation of the
a new family of compounds with a to describe complex systems, also affinity of binding between a ligand and
marked inhibitory activity. The joint thanks to the use of models quantome- a receptor would serve a day, test a
use of three-dimensional structures of chanical. billion compounds on the same
44 G Med Mil. 2013; 163(1): 31-48Puoi anche leggere

















































