Misure ripetute: un'introduzione - Universit'a degli Studi - Biostatistica Umg Catanzaro
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Università degli Studi
Magna Græcia di Catanzaro
UMG School of Phd Programmes
Life Sciences and Technologies
Misure ripetute: un’introduzione
www.biostatisticaumg.it
Valeria@Bonn
.. tenga presente che ogni
esperimento deve essere effettuato in
triplicato biologico ed in triplicato
tecnico (ad esempio: 3 Real-Time
PCR) su 3 replicati biologici dei
campioni ( = cellule con la
mutazione vs. cellule WT) ..
.. In questo caso la soluzione migliore
per analizzare i dati sembrerebbe
essere l’uso del T test perche‘
abbiamo 2 gruppi (Mutato vs WT)
ma in realta‘ i gruppi sono 6 (mutato
1/2/3 vs WT 1/2/3), dunque la
scelta dovrebbe ricadere su un
ANOVA?
Massimo Borelli www.biostatisticaumg.it
Autore
Massimo Borelli, Ph.D.
Anno Accademico 2017 – 2018
Contents
1 Introduzione. 1
1.1 il t test non va bene .. . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 .. la anova non va bene .. . . . . . . . . . . . . . . . . . . . . . . 3
1.3 .. e nemmeno la two-way anova non va bene. . . . . . . . . . . . 3
2 Lo strano caso delle gemelle Alice ed Ellen. 5
3 Le misure ripetute non sono indipendenti. 8
3.1 I modelli ad effetti fissi. . . . . . . . . . . . . . . . . . . . . . . . 8
3.2 Una simulazione ci fa scoprire il colpevole. . . . . . . . . . . . . . 9
4 I modelli ad effetti misti. 11
5 Approfondimento: perché manca il p-value? 13
5.1 Selezione con i criteri di informazione. . . . . . . . . . . . . . . . 13
5.2 Selezione con l’analisi della devianza. . . . . . . . . . . . . . . . . 14
5.3 Selezione con il parametric bootstrap. . . . . . . . . . . . . . . . . 15
1 Introduzione.
Valeria Lucchino pone una domanda molto concreta che è emersa presso i lab-
oratori del Deutsches Zentrum für Neurodegenerative Erkrankungen di Bonn.
Proviamo a simularla con un esempio buffo.
Abbiamo tre soggetti wild-type, a, b e c, e tre soggetti mutati, d, e ed f.
Dalle loro cellule abbiamo effettuato misure quantitative con la real time PCR,
ottenendo queste misure:
• soggetto a: 28, 29, 27
• soggetto b: 26, 25, 24
• soggetto c: 28, 29, 30
• soggetto d: 26, 27, 25
• soggetto e: 24, 23, 22
• soggetto f: 26, 27, 28
1
(Trucco: osservate che i valori di d, e ed f sono esattamente quelli di a, b e c
diminuiti di due, alla faccia dell’imprevedibilità.)
Esercizio 1 Create o con il foglio elettronico, oppure con R stesso, il dataset
che descriva questo buffo esperimento
rtpcr = c (28 , 29 , 27 , 26 , 25 , 24 , 28 , 29 , 30 , 26 , 27 , 25 , 24 ,
23 , 22 , 26 , 27 , 28)
soggetto = f a c t o r ( sort (rep( letters [1:6] , 3) ) )
gene = f a c t o r (c(rep( " wt " , 9) , rep( " mut " , 9) ) )
valeria = data.frame( rtpcr , soggetto , gene )
Listing 1: Un modo per creare con R il dataset buffo.
rtpcr topo gene
28 a wt
29 a wt
27 a wt
26 b wt
25 b wt
24 b wt
28 c wt
29 c wt
30 c wt
26 d mut
27 d mut
25 d mut
24 e mut
23 e mut
22 e mut
26 f mut
27 f mut
28 f mut
1.1 il t test non va bene ..
È vero, in questo esempio buffo abbiamo due gruppi, codificati dall’informazione
gene: mut e wt. Mettiamoci pure nelle (discutibilissime!) ipotesi di normalità ed
omoschedasticità dei dati provenienti dalle popolazioni wild-type e mutata, ed
eseguiamo il t test. Forse quel p-value appena appena significativo ci potrebbe
ringalluzzire, ed indurci a decidere che vi sia una differenza biologica tra i wild-
type ed i mutati. Ma c’è un problema gigantesco evidenziato in colore azzurro:
df = 16.
2df = 16 significa che abbiamo 16 gradi di libertà; sedici misure indipendenti
dalla media del gruppo dei mutati (25.3) e dalla media dei gruppo dei wild-type
(27.3).
Esercizio 2 Spiegate perché non siamo in presenza di dati indipendenti. In tal
caso, i gradi di libertà dovrebbero essere maggiori o minori? E di conseguenza,
cosa succederebbe all’intervallo di fiducia? Ed al p-value?
1.2 .. la anova non va bene ..
Se il t test non è giusto, allora andrà bene la Anova.. delusione:
La delusione è nascosta di nuovo in quei cinque gradi libertà, che stanno a
rappresentare l’indipendenza (questo è giusto!) dei dati di b da a, di c da a,
di d da a, eccetera. Ma abbiamo perduto per strada l’informazione relativa al
’cluster’, cioè gene. Peccato, perché qui il p-value era bellissimo, ed anche il
famigerato Referee # 2 ci sarebbe cascato per sempre ;-)
1.3 .. e nemmeno la two-way anova non va bene.
Per effettuare una two-way anova dobbiamo usare il comando generico del mod-
ello lineare, lm:
3Ma qui ci sono ancora molte cose che non vanno. Innanzitutto il p-value non
è cambiato rispetto all’anova del paragrafo precedente (’p = 0.0001178’), pure
dopo aver introdotto l’informazione gene. E poi osservate che il soggetto c non
differisce in senso statistico dal soggetto a. Ma supponiamo che per assurdo a
e b si scambino la maglietta tra di loro:
Non va proprio bene affatto, c’è sicuramente qualcosa di sbagliato: ma cosa?
Ed infine, per aggiungere ulteriore pepe alla faccenda: siamo d’accordo sul fatto
che Valeria ci assicura sia necessario effettuare tutto in triplicato. Ma se invece
di triplicato volessimo quadriplicare, o sestuplicare? Potremmo confrontare i
nostri risultati con quelli di Valeria? Leggiamolo nel secondo capitolo.
42 Lo strano caso delle gemelle Alice ed Ellen.
Le gemelle Alice ed Helen sono due anziane signore che, dopo aver condotto una
vita artistica di grande successo, decidono di riprendere gli studi di biostatistica
che avevano interrotto alcuni decenni fa. Alice ed Ellen decidono di fare uno
studio osservazionale: alzarsi dal letto assieme ogni mattina e immediata-
mente pesarsi, per rispondere alla seguente domanda: Alice ed Ellen hanno lo
stesso peso?
All’indomani, eseguito il primo esperimento e preso nota del responso della
bilancia (accuratissima, digitale, che non si lascia perturbare dalle onde gravi-
tazionali, ecc. ecc.) la situazione è la seguente:
Alice Ellen
73.60 73.80
A questo punto, Alice ed Ellen sarebbero propense a decidere che non hanno
lo stesso peso, giacché, ragionando da un punto di vista puramente matematico,
i due numeri non coincidono.
Ma le gemelle sanno che, nella Natura, la variabilità la fa da padrona[1] e cosı̀
scelgono di fare un secondo esperimento, ossia di pesarsi per cinque mattine
consecutive (studio osservazionale longitudinale, misure in ’quintuplicato
tecnico’):
Alice Ellen
1 73.60 73.80
2 73.40 73.50
3 74.10 74.60
4 73.50 73.80
5 73.20 73.60
Per dirimere la questione esse ricorrono al celebre test t di Student. Come tutti
ricordano, si vuole decidere se la media dei pesi di Alice sia diversa ’in senso
statistico’ dalla media dei pesi di Ellen, immaginando che per ciascuna di esse
siano stati osservati cinque numeri casuali provenienti da due variabili aleatorie
gaussiane, di media (nel senso di valore atteso, o speranza matematica) diversa
ma con la medesima dispersione (nel senso di deviazione standard, ovvero della
varianza).
Ecco qui di seguito il listato dei comandi per eseguire il test con R e l’output
fornito dal software.
5
alice = c (73.6 , 73.4 , 74.1 , 73.5 , 73.2)
ellen = c (73.8 , 73.5 , 74.6 , 73.8 , 73.6)
t . test ( alice , ellen , var. equal = TRUE )
Listing 2: Il test t con R
Two Sample t-test
data: alice and ellen
t = -1.2227, df = 8, p-value = 0.2562
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-0.865794 0.265794
sample estimates:
mean of x mean of y
73.56 73.86
Alice ed Ellen ora sarebbero propense a decidere che hanno lo stesso peso, in
base al p-value = 0.2562 (non inferiore al 5%). Infatti, la differenza tra il peso
medio di Alice (73.56) e quello di Ellen (73.86) dà luogo ad un consuntivo t =
-1.2227, il quale rispetto alla variabile aleatoria t di Student a df = 8 gradi di
libertà (5 pesi di Alice + 5 pesi di Ellen - 1 valor medio di Alice - 1 valor medio
di Ellen), equivale ad un’area di probabilità p pari a 0.2562, come vediamo nella
regione tratteggiata della figura sottostante.
0.4
densità di probabilità
0.3
0.2
0.1
0.0
-3 -2 -1 0 1 2 3
quantili della distribuzione t di Student
6Le gemelle tuttavia ricordano che l’affidabilità delle misure aumenta con il nu-
mero di repliche. Scelgono perciò di continuare a pesarsi complessivamente per
tre settimane, dando luogo al loro terzo esperimento. Riportiamo la tabella con
i dati grezzi dei pesi e il risultato del relativo test t di Student.
Alice Ellen Alice Ellen
1 73.60 73.80 12 74.10 74.60
2 73.40 73.50 13 73.60 73.80
3 74.10 74.60 14 73.40 73.60
4 73.50 73.80 15 74.10 74.40
5 73.20 73.60 16 73.50 73.70
6 74.00 74.40 17 73.20 73.50
7 73.60 73.80 18 74.00 74.40
8 73.30 73.50 19 73.60 73.90
9 74.20 74.30 20 73.30 73.60
10 73.60 73.90 21 74.20 74.50
11 73.40 73.60 - - -
Two Sample t-test
data: peso by gemella
t = -2.4594, df = 40, p-value = 0.01834
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-0.51183215 -0.05007261
sample estimates:
mean in group alice mean in group ellen
73.66190 73.94286
Colpo di scena! Alice ed Ellen ora si sentono confuse più che mai, perché
ora dovrebbero decidere che non hanno lo stesso peso, in base al p-value =
0.01834, significativo; contrariamente a quello che era accaduto nel secondo
esperimento. Tutto ciò è molto strano. Di chi è la colpa?
7Esercizio 3 Carmen De Caro ci segnala il paper apparso su Cell, primo nomne
Christine A. Olson, PMID: 29804833. Leggetelo e chiedetevi se nell’applicare
(Figura 1) la two-way anova gli autori non siano incorsi nell’errore spiegato nei
paragrafi 1.3 e 2 di questa dispensa.
3 Le misure ripetute non sono indipendenti.
Alice ed Ellen hanno commesso un errore: non si sono ricordate che il test t è
appropriato quando siamo in presenza di dati indipendenti e non, come in questo
caso, di dati correlati [7], come è tipico nei design sperimentali di tipo longi-
tudinale in cui si eseguono misure ripetute, in tempi successivi, sul medesimo
soggetto.
3.1 I modelli ad effetti fissi.
Vediamo la questione in maggior dettaglio. Il test t di Student è un modello
(statistico) lineare ad effetti fissi. Questo significa che detto µ = 73.66 il peso
medio di Alice ottenuto nel terzo esperimento, il peso medio di Ellen è superiore
a quello della gemella di una costante (effetto fisso) β2 = 0.28 = 73.94 − 73.66
(mentre per quello di Alice possiamo per completezza porre l’effetto fisso β1 =
0). E, di volta in volta, i pesi delle gemelle potrebbero essere perturbati da un
’rumore’ εij che varia, da gemella a gemella (i), e di giorno in giorno (j):
peso = µ + βi + εij
8I software riescono a stimare, matematicamente, il comportamento casuale del
’rumore’ εij , indicando la quantità che si chiama residual standard error. Vedi-
amolo con i comandi di R:
gemelle21 = read. csv ( f i l e . choose () , header = TRUE )
attach ( gemelle21 )
m o d e l l o e f f e t t i f i s s i = lm( peso ˜ gemella )
5 summary( m o d e l l o e f f e t t i f i s s i )
Listing 3: Il modello lineare ad effetti fissi con R
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 73.66190 0.08078 911.915terzo esperimento simulazione
74.6
12
3 11
21
16
74.4
6 15
18 6
9
74.2
EllenFinta
5 21
18 19
Ellen
7 3
74.0
14 8
2
73.8
10
19 20 1
4 713
1 12
10
16 13
9
73.6
73.4
5 20 11
14 17
15
17 8 2 4
73.2 73.4 73.6 73.8 74.0 74.2 73.0 73.4 73.8 74.2
Alice AliceFinta
Per intenderci, ricordiamo che al 21-esimo giorno i pesi di Alice ed Ellen er-
ano rispettivamente 74.20 e 74.50; lo vediamo evidenziato nell’angolo in alto a
destra del primo pannello. Il secondo pannello invece mostra una simulazione
casuale ottenuta partendo dai parametri stimati dal modelloeffettimisti che
abbiamo trovato. Come si vede, la nube di punti è del tutto caotica; e se provi-
amo a ripetere millanta volte questa simulazione, con il comando simulate
come vedete nel codice in basso, otterremo sempre una situazione disordinata
di questo genere, e praticamente mai come quella di sinistra. A sinistra, ci tro-
viamo in una situazione di elevata informazione; a destra, in una situazione di
assenza di informazione, ovvero di elevata entropia [3]. E questo contravviene
alla richiesta di adeguatezza di un modello statistico [4, 6], che potremmo in
maniera naı̈ve esprimere in questo modo:
Un modello statistico M è adeguato a descrivere i dati osservati a
priori, rispetto ad un modello peggiore M̂ , se, generando a posteriori
per mezzo del modello M nuovi dati in maniera casuale, questi ultimi
’esibiscano una grande verosimiglianza’ rispetto ai dati originali. [5]
Alice = peso [ gemella == " alice " ]
Ellen = peso [ gemella == " ellen " ]
par( mfrow = c (1 , 2) )
plot ( j i t t e r ( Alice ) , j i t t e r ( Ellen ) , xlab = " Alice " , ylab = "
Ellen " )
5 simulazione = u n l i s t ( simulate ( m o d e l l o e f f e t t i f i s s i ) )
AliceFinta = simulazione [1:21]
EllenFinta = simulazione [22:42]
plot ( AliceFinta , EllenFinta )
Listing 4: Simulazione con un modello ad effetti fissi
104 I modelli ad effetti misti.
Ritorniamo dunque all’esempio buffo del capitolo 1. Abbiamo provato a con-
durre l’analisi statistica applicando tre modelli ad effetti fissi, tutti e tre
sbagliati:
• t test: rtpcr ∼ gene
• one-way anova: rtpcr ∼ soggetto
• two-way anova: rtpcr ∼ soggetto + gene
Abbiamo invece bisogno di un quadro teorico in cui si riesca a specificare il
fatto che gene rappresenta una variabile di gruppo (effetto fisso), sulla quale
vogliamo cercare differenze di popolazioni; mentre soggetto è una variabile
su cui si replicano – quante volte si vogliono – le misure (effetto casuale).
Abbiamo dunque bisogno di una formula che combini tra loro un effetto fisso
ed un effetto casuale per spiegare i valori della real time PCR:
• modello ad effetti misti: rtpcr ∼ gene + (1|soggetto)
Il pacchetto lme4 [2] rappresenta attualmente la migliore risorsa per questo tipo
di indagini. Vediamo come si fa, utilizzando il comando lmer:
l i b r a r y ( lme4 )
relazione = rtpcr ˜ gene + (1| soggetto )
modello = lmer ( relazione )
summary( modello )
Listing 5: l’analisi della varianza con misure ripetute utilizzando i modelli ad
effetti misti.
Otteniamo un output ricchissimo di informazioni. Ma quello che salta immedi-
atamente all’occhio è che ’non ci sono le stelline’, non ci sono dei p-value. E se
11questo può sconcertare il neofita, invece questo conforta lo statistico che sa di
aver vissuto in un periodo di profonda crisi scientifica, per aver abusato in letter-
atura di questo strumento. Vediamo in dettaglio la parte di nostro immediato
interesse:
Riscopriamo come già sapevamo che in media i mutati hanno un valore di
rtpcr pari a 25.3, mentre i wt hanno un incremento medio di 2 (ossia 27.3,
come visto con il t test del capitolo 1). Tuttavia la banda di fiducia che viene
individuata dallo standard error (inteso proprio nel vero senso del termine: la
deviazione standard della media campionaria), o equivalentemente il consuntivo
t = 1.177, ci fa capire che ’non c’è differenza significativa’ tra le due medie,
contrariamente a quello che il capitolo 1 ci lasciava intendere. Per capire meglio
questa frase, osserviamo questo disegno:
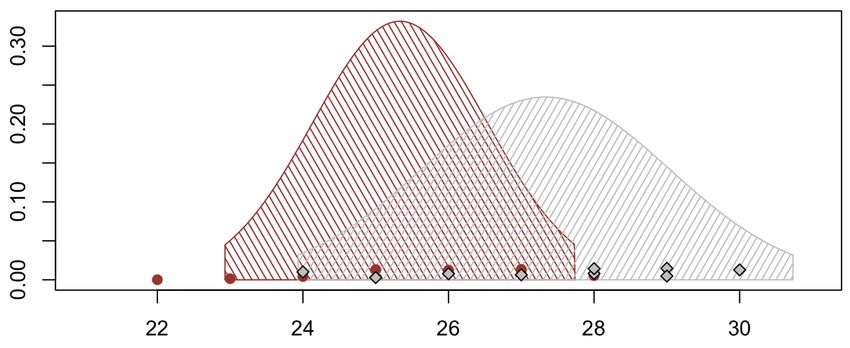
In orizzontale, i pallini rossi ed i diamanti grigi rappresentano i valori rtpcr
mutati e wild - type leggermente perturbati. Il punto di massimo della gaussiana
rossa (25.3) cade nella regione tratteggiata grigia, che è individuata dalla media
grigia (27.3) aumentata e diminuita di due deviazioni standard (± 2 · 1.7). E
viceversa, il punto di massimo della distribuzione grigia cade nella fascia rossa.
Quindi siamo propensi a decidere appunto che nell’esperimento buffo non vi sia
un effetto genetico tra i mutati ed i wild - type in base alle letture della rtpcr.
Esercizio 4 Alla luce di quanto appena visto, avete ancora fiducia sul fatto che
il paper segnalatoci da Carmen De Caro (Christine A. Olson, PMID: 29804833)
giunga a conclusioni supportate dalla analisi statistica appropriata?
125 Approfondimento: perché manca il p-value?
Nella figura precedente abbiamo traggeggiato due gaussiane al di sopra dei valori
misurati di rtpcr. Ebbene, questo è un errore. Infatti, Douglas Bates [2] spiega
come sia praticamente impossibile disporre di una valida teoria matematica che
determini il grado di libertà delle distribuzioni statistiche inerenti i disegni di
ricerca con misure ripetute. Quindi, non possiamo disporre di un calcolo esatto
per determinare il p-value. Come possiamo ovviare a questo inconveniente? In
due modi:
1. selezionando i modelli in base ai criteri di informazione
2. selezionando i modelli in base alla analisi della devianza
3. selezionando i modelli con il parametric bootstrap.
I primi due metodi sono validi nella circostanza in cui si vogliano testare modelli
con diversi effetti fissi ma con medesimo effetto casuale [5], mentre il terzo
metodo (che però ha lo svantaggio di essere computazionalmente intensivo e
richiede qualche capacità di programmazione) è sempre valido.
5.1 Selezione con i criteri di informazione.
Se i mutati ed i wild - type fossero effettivamente diversi in base al gene, il
modello adeguato a descriverli sarebbe:
• modello gene: rtpcr ∼ gene + (1|soggetto)
Ma se al contrario il gene non avesse effetto su rtpcr dovremmo preferire il
cosiddetto ’modello nullo’:
• modello nullo: rtpcr ∼ 1 + (1|soggetto)
Ora c’è da sapere un dettaglio importante: la stima dei parametri nei mod-
elli ad effetti misti avviene utilizzando il cosiddetto criterio ristretto di mas-
sima verosimiglianza (in inglese, restricted maximum likelihood ), ’REML’. Ma
se vogliamo calcolare il valore del criterio di informazione di Akaike associato ad
un modello ad effetti misti, dobbiamo stimare i parametri col citerio di massima
verosimiglianza, il che comporta delle leggere - ma non trascurabili - variazioni
sugli standard error. Ecco qui di seguito la sintassi ed il responso:
13
relazionegene = rtpcr ˜ gene + (1| soggetto )
rela zionenul la = rtpcr ˜ 1 + (1| soggetto )
modellogene = lmer ( relazionegene , REML = FALSE )
5 modellonullo = lmer ( relazionenulla , REML = FALSE )
AIC ( modellogene )
AIC ( modellonullo )
Listing 6: la selezione tra due modelli ad effetti misti (con il medesimo effetto
casuale) per mezzo dei criteri di informazione.
Siccome nell’interpretare i criteri di informazione vale il paradigma del minore
caos possibile, please!, il modello nullo è da preferire, e quindi ri-concludiamo
che nell’esperimento buffo non vi sia un effetto genetico tra i mutati ed i wild -
type in base alle letture della rtpcr.
Approfondimento. È un dettaglio importante quello della stima in mas-
sima verosimiglianza. Se non ci ricordassimo di farlo (default: REML = TRUE)
potremmo persino giungere alla conclusione errata:
5.2 Selezione con l’analisi della devianza.
Si tratta di un metodo praticamente equivalente a questo appena visto, che però
ha un vantaggio, che riempie di gioia i referee svogliati, i quali non desiderano
studiare come funzionino i mixed models, ma che pretendono a tutti i costi
di vedere con i loro occhi un p-value. Si procede utilizzando il comando della
analisi della devianza, anova:
14
relazionegene = rtpcr ˜ gene + (1| soggetto )
rela zionenul la = rtpcr ˜ 1 + (1| soggetto )
modellogene = lmer ( relazionegene , REML = FALSE )
5 modellonullo = lmer ( relazionenulla , REML = FALSE )
anova( modellogene , modellonullo )
Listing 7: la selezione tra due modelli ad effetti misti (con il medesimo effetto
casuale) per mezzo della analisi della devianza.
Il p-value 0.18 ci fa dedurre che il modello nullo ed il modello gene non dif-
feriscano. Pertanto il modello nullo è da preferire perché possiede 3 gradi di
libertà, mentre il modello gene è più costoso in quanto ne richiede quattro.
Quindi ri-ri-concludiamo che nell’esperimento buffo non vi sia (p = 0.18) un
effetto genetico tra i mutati ed i wild - type in base alle letture della rtpcr, per
la gioia del referee ancorato agli ’old good days’.
5.3 Selezione con il parametric bootstrap.
Si tratta di un argomento un pochino complesso per chi lo vede per la prima
volta, e lo trovate descritto a pagina 18 di questa dispensa: http://www.dmi.
units.it/pubblicazioni/Quaderni_Didattici/56_2011.pdf, oppure diret-
tamente in [5]. In parole semplici (e la simulazione del listing 4 è basata proprio
su quel procedimento) si tratta di genereare come dicevamo a pagina 10 dei nu-
meri casuali con il modello statistico ’più ricco’ e vedere se essi vengono ’spiegati
bene’ anche dal modello più spartano, o no.
References
[1] Naomi Altman and Martin Krzywinski. Points of significance: Sources of
variation. Nature methods, 12(1):5–6, 2015.
[2] Douglas Bates, Martin Maechler, Ben Bolker, and Steven Walker. lme4:
Linear mixed-effects models using Eigen and S4, 2014. R package version
1.1-7.
[3] Kenneth P Burnham and David R Anderson. Model selection and multimodel
inference: a practical information-theoretic approach. Springer Science &
Business Media, 2003.
15[4] Michael J Crawley. Statistics: an introduction using R. John Wiley & Sons,
2005.
[5] Julian J Faraway. Extending the linear model with R: generalized linear,
mixed effects and nonparametric regression models. CRC press, 2005.
[6] John K. Kruschke. Doing Bayesian data analysis: A tutorial with R, and
BUGS. Academic Press, 2011.
[7] Geert Verbeke and Geert Molenberghs. Linear mixed models for longitudinal
data. Springer Science & Business Media, 2000.
16Puoi anche leggere

















































