La Rivista Italiana delle Malattie Rare - La Rivista delle Malattie Rare
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù

ISSN: 2612-2588
Rare Disease Day®
la Rivista
La Rivista Italiana delle Malattie Rare
review il caso clinico pagina
anno V
La malattia di Whipple Malattia di Fabry con dismorfologica
febbraio La sclerosi sistemica cardiomiopatia ipertrofica Sindrome CDKL5
2021 Una insufficienza cardiaca e sindromi Rett-like
l’opinione
ad eziologia sospetta
1
numero La pediatria della
disabilità: tutti convocati
QUADRIMESTRALE DI ATTUALITA’ IN MEDICINA Pubblicazione registrata al Tribunale di Milano
n. 11 del 10 gennaio 2017 - Poste Italiane Spa Spedizione in Abbonamento Postale - 70% - LO/MI
2

la Rivista
sommario La Rivista Italiana delle Malattie Rare
editoriale
Chiari-scuri di quest’anno horribilis B. Bembi 5
review
La malattia di Whipple M.L. Nicolardi, F. Biagi 6
La sclerosi sistemica S. Guiducci 9
l’opinione
La pediatria della disabilità: tutti convocati A. Selicorni, G. Zampino 13
il caso clinico
Malattia di Fabry con cardiomiopatia ipertrofica
L. Roggero, F. Pieruzzi 15
Una insufficienza cardiaca ad eziologia sospetta
S. Perlini, F. Musca 18
pagina dismorfologica
Sindrome CDKL5 e sindromi Rett-like A. Vignoli, I. Viganò, A. Peron 21
il farmaco
Patisiran: la prima terapia RNAi per il trattamento dell'amidoilosi ereditaria mediata
dalla transtiretina A. Esposito, C. Panico, M. Medaglia
24
letteratura
Coinvolgimento cardiologico nella malattia di Fabry: revisione della letteratura
E. Lonni, P. Lusardi, M. Cannillo, W. Grosso Marra 26
L'impiego dell'imaging nella diagnosi e nella gestione dell'amiloidosi cardiaca L. Obici 28
dalle reti europee
ERN-EuroBloodNet, la Rete di Riferimento Europea
per le malattie ematologiche rare L. Barcella, A. Falanga 30
la voce delle associazioni
La transizione nelle malattie rare: il punto di vista di pazienti e famiglie R. Lala 32
3
direttore
scientifico
Bruno Bembi
Medico Pediatra e Genetista, Trieste
comitato
di redazione
Simone Baldovino, CMID, Centro di Coordinamento Rete Interregionale Malattie Rare Piemonte e
Valle d’Aosta, Ospedale S. Giovanni Bosco – ASL Città di Torino; Dipartimento di Scienze Cliniche e
Biologiche, Università di Torino
Daniela Concolino Responsabile Centro Regionale Pediatria Genetica e Malattie Rare, Università
degli Studi “Magna Græcia”, Catanzaro
Daniela Damiani Professore Associato di Ematologia, Dipartimento di Scienze Mediche Sperimentali
e Cliniche, Azienda Sanitaria Universitaria Integrata di Udine
Erica Daina Responsabile Laboratorio di Documentazione e Ricerca sulle Malattie Rare, IRCCS -
Istituto di Ricerche Farmacologiche Mario Negri, Bergamo
Andrea Elena Dardis Responsabile Laboratorio, Centro di Coordinamento Regionale per le Malattie
Rare, Azienda Sanitaria Universitaria Integrata di Udine
Emanuela De Juli Responsabile Coordinamento Aziendale Malattie Rare, Struttura complessa di
Pneumologia, ASST Grande Ospedale Metropolitano Niguarda, Milano
Massimo Medaglia Direttore S.C. Farmacia, ASST Grande Ospedale Metropolitano Niguarda, Milano
Laura Obici Dirigente Medico I livello, Centro per lo Studio e la Cura delle Amiloidosi Sistemiche,
Fondazione IRCCS Policlinico San Matteo, Pavia
Rossella Parini Consulente medico-scientifico per la Fondazione Monza e Brianza per il Bambino e
la sua Mamma, Struttura Semplice Malattie Rare, Clinica Pediatrica, Ospedale San Gerardo, Monza
Dario Roccatello CMID Centro di Ricerche di Immunopatologia e Documentazione su Malattie
Rare, Coordinamento Interregionale Rete Malattie Rare del Piemonte e della Valle d’Aosta, SCDU
Nefrologia e Dialisi, Ospedale San Giovanni Bosco e Università di Torino
Angelo Selicorni Direttore Unità Operativa Complessa di Pediatria, ASST-Lariana, Como
collaboratori
Generoso Andria, Napoli; Maurizio Aricò, Bari; Maurizio Averna, Palermo; Tommaso Beccari, Perugia;
Andrea Bordugo, Verona; Marco Confalonieri, Trieste; Carlo Dionisi Vici, Roma; Vincenzo Leuzzi,
Roma; Tiziana Mongini, Torino; Giancarlo Parenti, Napoli; Luca Sangiorgi, Bologna; Gioacchino
Scarano, Benevento; Maurizio Scarpa, Udine; Antonio Toscano, Messina; Giuseppe Zampino, Roma
norme per gli autori
la Rivista
I contributi spontanei (titolo e scaletta contenutistica) dovranno essere inviati esclusivamente
via e-mail, alla Segreteria di Redazione di MR (arianna.nespolon@medpointsrl.it). Il Comitato
di Redazione si riserva di valutarne la pubblicazione sulla testata, dandone pronto riscontro
La Rivista Italiana delle Malattie Rare
all’Autore.
per richiedere MR
la Rivista
Volete richiedere la Rivista Italiana delle Malattie Rare? Collegandovi alla sezione dedicata
4 del sito http://www.malattierare.eu/pages/richiediRivista potrete accedere al modulo da
La Rivista Italiana delle Malattie Rare compilare per ricevere gratuitamente la rivista al vostro indirizzo.
editoriale
Chiari-scuri di quest’anno
Bruno Bembi Medico
Pediatra e Genetista, Trieste horribilis
C ari lettori e cari amici, è passato un anno dal mo-
mento in cui, nell’iniziale disattenzione, bussava
alle porte del mondo questa terribile pandemia. Al
Translational Myology. I due clinici auspicano l’utilizzo
di questa app, che ogni paziente può scaricare sul
proprio smartphone, anche per altre patologie di am-
momento in cui questo editoriale viene scritto i dati ci bito neuromuscolare.
dicono che si è portata via 2.665.354 vite ed ha colpi- In passato esperienze simili, il più delle volte sostenu-
to più di 104 milioni di abitanti del pianeta. Un pensie- te da aziende farmaceutiche, sono state tentate nel
ro va agli anziani che hanno concluso da soli la loro campo di altre patologie metaboliche, ma con scar-
strada, a quei lavoratori della sanità e dell’assistenza so successo. Un insuccesso dovuto in molti casi sia ad
che sono stati sconfitti mentre erano impegnati nella una scarsa dimestichezza dei pazienti e medici con i
quotidiana e silenziosa battaglia per aiutare gli altri, a mezzi informatici, sia alla facilità di accedere ai servizi
quei giovani e ai bambini che inaspettatamente han- assistenziali e ai centri clinici esperti.
no visto chiudersi i propri sogni. Un grazie a medici e Come la storia ci insegna, i momenti di crisi aprono
scienziati che ci hanno ridato una speranza per il futu- sempre nuove vie, in questo caso la diffusione dell’in-
ro, ma anche a quelle aziende che in pochi mesi sono formation technology (IT) è stata una necessità ed un
riuscite a realizzare i vaccini che ci salveranno. Loro momento di progresso per il nostro Paese. Lanciamo
chiediamo un ulteriore impegno etico, di garantire a un sasso: un’analisi sugli strumenti IT rivolti ai pazienti
tutti i cittadini del mondo l’accesso al vaccino. per la gestione quotidiana delle malattie rare potreb-
La quantità di articoli scientifici pubblicati in questi 12 be essere un campo d’indagine da approfondire ed
mesi sul tema COVID-19 è impressionante, si fa fatica utile alla comunità. I ricercatori di sicuro saranno sen-
a seguire la letteratura ed in particolare il suo valo- sibili, sarebbe auspicabile che altrettanto sensibili a
re, garantito dal percorso di peer review. Carente in raccogliere questo stimolo possano essere i decision
questo contesto è però un tentativo d’analisi ed un maker in politica sanitaria.
bilancio di quello che la pandemia ha voluto dire per Rimanendo nel campo dell’IT, il 2020 è stato un anno
i malati rari: poco esiste. Alla preoccupazione iniziale positivo per la nostra rivista. Il sito web è stato visitato
di pazienti, famiglie e curanti apparentemente non è complessivamente da 82.211 utenti individuali, con un
corrisposta una ripercussione negativa sull’erogazio- aumento del 40% di download rispetto all’anno pre-
ne delle cure ed il mantenimento degli standard as- cedente, che si è tradotto in più di 7000 pubblicazioni
sistenziali adeguati. scaricate. Un ulteriore elemento di positività è osser-
La situazione di stress e di rischio ha stimolato nuo- vare che lo sforzo di diffusione fatto per toccare tutte
ve modalità di lavoro, sperimentate con successo le aree del Paese sta dando i suoi frutti. Prevalgono
nell’industria e nella scuola ed ha portato a matura- sempre le visite provenienti da aree geografiche del
re l’applicazione della telemedicina, o meglio delle nord Italia, 46,6%, ma a differenza del passato la mag-
cosiddette app mediche, nella cura dei malati rari, gioranza complessiva dei contatti arriva dalle altre re-
che rimarranno certamente utili anche ad emergen- gioni: 32,2% da quelle del Centro e 21,3% da quelle
za finita. del Sud.
Un esempio è lo sviluppo della AGIkit-app, risultato di Una criticità rimane la prevalenza nel contributo alla
un lavoro congiunto tra l’Associazione Italiana Glico- realizzazione scientifica della rivista di articoli proposti
genosi e l’Associazione Italiana di Miologia. Si tratta di da ricercatori e centri delle aree settentrionali. Questa
uno strumento che collega pazienti e centri di cura e consapevolezza rafforza il nostro impegno a coinvol-
permette di intervenire in situazioni di difficoltà clini- gere le eccellenze presenti su tutto il territorio naziona-
ca ed assistenziale. Il risultato è stato recentemente le, ben consci della ricchezza della ricerca scientifica
illustrato alla comunità scientifica internazionale dai e dell’impegno clinico-assistenziale esistente.
professori Corrado Angelini e Gabriele Siciliano in un
loro recente lavoro apparso su European Journal of Bruno Bembi
5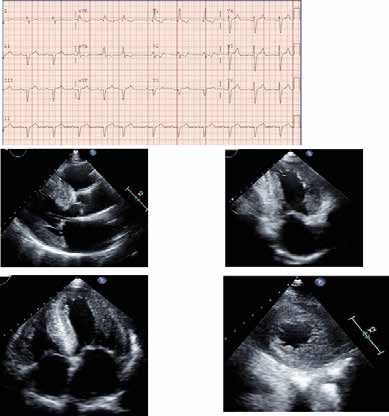
review
Maria Luisa Nicolardi, Federico Biagi
Istituti Clinici Scientifici Maugeri, IRCSS, Unità di Gastroenterologia dell’Istituto di Pavia, Università di Pavia
La malattia di Whipple
La malattia di Whipple (MW) è una DRB1*06, oltre ad essere descritti casi Il TW, inoltre, induce nei monociti infetti
rara patologia cronica ad interessa- familiari in letteratura [3]. l’espressione di IL-16 il che ne inibisce
mento sistemico, caratterizzata da la maturazione. Ciò determina una ri-
una presentazione clinica aspecifica Patogenesi sposta immunitaria prevalentemente
ed eterogenea che, se non pronta- L’infezione primaria, probabilmente antinfiammatoria (Th2). La risposta Th1
mente riconosciuta e trattata, può contratta nell’infanzia con trasmis- è invece ridotta come pure la pro-
essere fatale [1]. È una malattia rara, sione interumana, tende a decorrere duzione di immunoglobuline contro il
con una prevalenza di 1-3/1.000.000 asintomatica o a dare una gastroen- TW. Pur fagocitando il batterio, i ma-
e un’incidenza annuale
la Rivista
La Rivista Italiana delle Malattie Rare
soppressiva durante la fase prodromi- mente. Può manifestarsi sia con lesioni
tab. 1 Manifestazioni cliniche
ca. In questo stadio, inoltre, possono cerebrali multiple, identificabili tramite
e prevalenza
manifestarsi febbre, linfoadenopatie, TC o RMN e responsabili di segni e
anemia, iperpigmentazione cutanea Calo ponderale >90% sintomi neurologici molto eterogenei,
ed essere interessati organi quali cuo- Steatorrea 90% che con una singola lesione cerebra-
re, polmoni, pelle, ossa e muscoli (Tab. Ipoalbuminemia 90% le con conseguenti deficit neurologici
1). Anemia 80-85%
focali.
Un coinvolgimento del SNC, presente
Diarrea 80%
nel 10-40% dei pazienti, può verificarsi Diagnosi
Artralgie 67-80%
in caso di diagnosi tardiva ed è il prin- La diagnosi di MW classica si basa
cipale responsabile di una prognosi Linfoadenopatie 55-60% sul riscontro nei tessuti affetti, in par-
infausta. Le manifestazioni neurolo- Dolore addominale 45-55% ticolare nelle biopsie duodenali, di
giche sono estremamente eteroge- Iperpigmentazione cutanea 40-45% macrofagi schiumosi diastasi resisten-
nee, nonostante la cefalea e i disturbi Febbre 35% ti positivi alla colorazione con acido
cognitivi rappresentino i sintomi più periodico di Schiff (PAS) e negativi
Manifestazioni neurologiche 10-40%
frequenti; i disordini del movimento alla colorazione di Ziehl-Neelsen (Fig.
Manifestazioni oculari 6-8%
oculare, come l’oftalmoplegia sopra- 1). Nei macrofagi si osservano inclu-
nucleare progressiva o la mioaritmia sioni citoplasmatiche granulari PAS
oculofacioscheletrica, sono invece positive. I villi intestinali sono tozzi e
Manifestazioni
considerati patognomonici (Tab. 2). tab. 2 neurologiche della MW accorciati e presentano della lin-
Nel 50% dei pazienti, tuttavia, l’interes- fangiectasie secondarie all’infiltrato
samento neurologico è asintomatico Atassia Crisi epilettiche macrofagico. È inoltre possibile ot-
generalizzate
e può essere rilevato solo mediante tenere una diagnosi biomolecolare
l’esecuzione di una reazione a cate- Dismetria Inattenzione della malattia mediante tecnica PCR
na della polimerasi (PCR) per TW su Emiplegia Cefalea che ricerca specificamente il TW. Dal
liquor. Oltre alla sopradescritta forma Ipoestesia a guanto/calza Alterazioni cognitive momento che nel 50% dei casi si può
di Whipple, definita classica, esistono Segno di Babinski Sindrome amnesica
avere un interessamento neurologico
anche due forme particolari di MW, asintomatico, è opportuno sottoporre
Iperreflessia Demenza
meno frequenti nella pratica clinica, a PCR per TW su liquor tutti i pazienti
Mioaritmia Disartria
di cui sono stati descritti alcuni casi in oculofacioscheletrica
con recente diagnosi di MW [2,5,6].
letteratura: l’endocardite associata al
Oftalmoplegia Afasia
TW e il MW ad interessamento neuro- sopranucleare progressiva Terapia
logico isolato. L’endocardite associa- Disordini oculari verticali Irritabilità
La terapia antibiotica induce un ra-
ta a TW si manifesta senza un coin- pido miglioramento clinico e una
Nistagmo Aggressività
volgimento di altri organi o apparati. remissione duratura nella maggior
Diplopia Insonnia
Il TW è uno degli agenti eziologici più parte dei pazienti, sebbene possano
frequentemente riscontrati nelle en- Papilledema Ipersonnia permanere sintomi neurologici; è fon-
docarditi con esame colturale nega- Deficit del VII nervo cranico Polidipsia damentale, quindi, l’utilizzo di antibio-
tivo. In questi casi, i criteri di Duke per Meningoencefalite tici in grado di oltrepassare la barriera
la diagnosi di endocardite spesso non ematoencefalica (BEE). La terapia at-
vengono soddisfatti e la diagnosi si tualmente raccomandata prevede
basa sull’analisi istologica o sulla PCR Un coinvolgimento del SNC, l’utilizzo di ceftriaxone o meropenem
della valvola cardiaca espiantata, più principale responsabile di per via endovenosa per due settima-
frequentemente la valvola aortica. La ne, seguita dall’assunzione orale di
MW a interessamento neurologico
una prognosi infausta, può trimetoprim-sulfametossazolo per al-
isolato, molto rara, ha una prognosi in- verificarsi in caso di diagno- meno un anno [1,7]. Sebbene questa
fausta se non diagnosticata precoce- si tardiva terapia abbia un’eccellente risposta
La malattia di Whipple M.L. Nicolardi, F. Biagi 7
clinica, non è chiaro se prevenga re- maci immunosoppressivi prima del-
Biopsia duodenale
cidive a lungo termine, di cui sono fig. 1 al microscopio ottico: la corretta diagnosi di MW. In questi
stati descritti casi in letteratura. Per- MW alla diagnosi pazienti, dopo aver verificato l’avve-
tanto, altri autori suggeriscono una nuta eradicazione del TW mediante
terapia con idrossiclorochina e doxi- PCR, va tempestivamente associata
ciclina per un anno, seguita poi da anche una terapia corticosteroidea
profilassi a vita con doxiciclina [5,7]. [9,10]. La IRIS è una condizione che
può essere infatti fatale. Infine, tra le
Follow-up complicanze possiamo annoverare
Il follow-up prevede indagini strumen- anche le recidive di malattia, soste-
tali inizialmente a 6 e 12 mesi, poi an- nute dalla grande diffusione del TW
nualmente per i primi 3 anni e, infine, nell’ambiente e dalla predisposizione
ogni 3 anni per il resto della vita [1]. genetica dei pazienti. Sebbene que-
Nei macrofagi si osservano inclusioni citoplasmatiche
Nei pazienti con manifestazioni ga- granulari PAS positive. I villi intestinali sono tozzi e ste si presentino più comunemente
stroenteriche è indicata la gastro- accorciati e presentano dalla linfangiectasie subito dopo la sospensione della te-
secondarie all’infiltrato macrofagico.
scopia con biopsie duodenali con rapia antibiotica, possono verificar-
colorazione PAS per un monitoraggio La diagnosi si basa sul ri- si anche molti anni dopo la fine del
istologico; sebbene l’infiltrato macro- trattamento. Anche se queste com-
scontro nei tessuti affetti di
fagico rimanga PAS positivo per anni, plicanze non sono molto frequenti,
il suo aspetto cambia in modo carat-
macrofagi schiumosi dia- il loro impatto sulla morbidità e sulla
teristico in risposta alla terapia anti- stasi resistenti PAS positivi e prognosi a lungo termine dei pazienti
biotica. La PCR specifica per TW, inve- Ziehl-Neelsen negativi con MW è negativo.
ce, si negativizza più rapidamente ed
è un metodo estremamente valido Conclusioni
per il follow-up dei tessuti interessati. pia della MW, il coinvolgimento cere- La MW è una condizione molto rara,
brale era considerato la complicanza ma fatale se non prontamente rico-
Complicanze più severa, a prognosi infausta. nosciuta e trattata. Poiché le sue ca-
Alcuni pazienti possono sviluppare Nell’ultimo decennio è emersa l’im- ratteristiche cliniche sono aspecifiche
complicanze, quali un coinvolgimen- portanza dell’IRIS, un processo infiam- ed eterogenee ed interessano diversi
to neurologico, la sindrome infiamma- matorio aspecifico che si presenta nel organi, la MW deve essere tenuta in
toria da immunoricostituzione (IRIS) o 10% dei pazienti, principalmente con considerazione non solo da gastroen-
le recidive di malattia [2,5,8]. febbre o artralgie, dopo l’inizio della terologi, infettivologi, internisti, reuma-
Prima dell’introduzione di antibiotici in terapia antibiotica. Fattore di rischio tologi, neurologi, cardiologi, ma da
grado di superare la BEE come tera- per lo sviluppo di IRIS è l’utilizzo di far- tutti i medici.
Bibliografia
1. Marth T, Moos V, Müller C, et al. Tropheryma whipplei infection and Whipple's disease. Lancet Infect Dis. 2016; 16:13-22.
2. Schneider T, Moos V, Loddenkemper, C et al. Whipple’s disease: new aspects of pathogenesis and treatment. Lancet Infect Dis. 2018; 8:179-90.
3. Marth T, Raoult D. Whipple's disease. Lancet. 2003; 361(9353):239-46.
4. Marth T, Kleen N, Stallmach A, et al. Dysregulated peripheral and mucosal Th1/Th2 response in Whipple’s disease. Gastroenterology. 2002; 123:1468–1477.
5. Fenollar F, Puéchal X, Raoult D. Whipple’s disease. N Engl J Med. 2007; 365:55-66.
6. Von Herbay A, Ditton HJ, Maiwald M. Diagnostic application of a polymerase chain reaction assay for the Whipple’s disease bacterium to intestinal
biopsies. Gastroenterology. 1996; 110:1735-43.
7. Biagi F, Biagi GL, Corazza GR. What is the best therapy for Whipple's disease? Our point of view. Scand J Gastroenterol. 2017; 52:465-466.
8. Lagier JC, Fenollar F, Lepidi H, et al. Failure and relapse after treatment with trimethoprim/sulfamethoxazole in classic Whipple's disease. J Antimicrob
Chemother. 2010; 65:2005-12.
9. Feurle GE, Moos V, Schinnerling K, et al. The immune reconstitution inflammatory syndrome in whipple disease: a cohort study. Ann Intern Med. 2010;
153:710-7.
10. Biagi F, Trotta L, Di Stefano M, et al. Previous immunosuppressive therapy is a risk factor for immune reconstitution inflammatory syndrome in Whipple's
disease. Dig Liver Dis. 2012; 44:880-2.
8 MR La Rivista Italiana delle Malattie Rare anno V - n. 1 - febbraio 2021
review
Serena Guiducci
Direttore SOD Reumatologia, Responsabile Scleroderma Unit, Dipartimento Medicina Sperimentale e Clinica, Azienda Ospedaliera Careggi,
Università degli Studi di Firenze
La sclerosi sistemica
La sclerosi sistemica (SSc) o sclero- con disregolazione del tono vasco- ti per la SSc suggerisce che le cause
dermia, ovvero “pelle indurita”, dal lare, clinicamente evidente come di questa malattia siano da attribuire
greco σκληρός (duro) e δέρμα (pelle) è fenomeno di Raynaud (FR), le “mani principalmente a fattori epigenetici,
una malattia cronica rara del tessu- fredde” in oltre il 90% dei casi. Tali piuttosto che genetici. In particola-
to connettivo, ad eziologia multifat- anormalità microcircolatorie sono re, analizzando i profili di metilazione
toriale, caratterizzata da alterazioni le prime manifestazioni della SSc e del DNA di linfociti da sangue perife-
del sistema immunitario con produ- possono precedere di mesi o anni il rico di gemelli monozigoti discordan-
zione di autoanticorpi, disfunzione coinvolgimento cutaneo e viscera- ti per la malattia, sono state osserva-
endoteliale e progressivo accumulo le. L’intero processo è caratterizzato te differenze solo nei geni presenti sul
di tessuto fibroso a carico della cute da perdita dei capillari, con severe cromosoma X. La SSc ha una predo-
e degli organi interni: apparato ga- manifestazioni di ischemia periferica minanza femminile con un rapporto
stroenterico, cuore, polmone, rene. (soprattutto a livello delle mani) con tra i due sessi di circa 12:1, il genere
La prima descrizione risale al XVIII se- formazione di ulcere digitali e gan- può quindi avere un ruolo determi-
colo da parte del medico Curzio da grena (fino all’autoamputazione di nante nello sviluppo di tale malattia.
Napoli, ma il termine “sclerodermia” falangi), causando così grave disa- L’esordio avviene più frequentemen-
è molto più recente. L’accumulo di bilità ai pazienti. Definita da W. Osler te fra i 20 ed i 50 anni e l'incidenza
tessuto fibroso a carico degli orga- “the most terrible of all human disea- è stimata tra i 4 e i 20 nuovi casi per
ni rende la malattia invalidante con ses”; è infatti terribile, per il paziente, 1.000.000 per anno.
conseguente compromissione della assistere all’avanzare della malattia,
qualità di vita del paziente. La sclerosi che altera i lineamenti del viso e Segni clinici
cutanea e l’atrofia muscolare carat- le espressioni mimiche, diminuisce L’elemento caratterizzante la SSc
teristiche della malattia causano se- le capacità funzionale delle mani che inizia con “le mani fredde”, è
veri cambiamenti nell’aspetto fisico, dove compaiono ulcere cutanee però il progressivo ispessimento ed
con un inevitabile impatto sulla sfera dolorose e necrotiche, limita l’au- indurimento della cute e del tessu-
emotiva e sul benessere psicologico tosufficienza del paziente e altera, to connettivo sottocutaneo, la cui
del paziente. Inoltre la SSc è caratte- inoltre, importanti funzioni d’organo. estensione (valutata mediante il Ro-
rizzata da alterazioni microvascolari dnan Skin Score) consente di distin-
Patogenesi guere classicamente una forma limi-
Le cause non sono ancora state tata, (interessamento delle regioni
identificate e i numerosi studi sulla distali degli arti superiori ed inferiori
La SSc è caratterizzata da
genetica nella sua patogenesi non ed eventualmente del volto), ed una
alterazioni del sistema im- hanno portato a risultati esaustivi. Il forma diffusa, che colpisce anche
munitario, disfunzione endo- fatto che gemelli monozigoti, ossia le regioni prossimali degli arti e del
teliale e progressivo svilup- caratterizzati dallo stesso patrimonio tronco (Tab. 1).
po di fibrosi genetico, possano essere discordan- Le principali cause di morte sono
La sclerosi sistemica S. Guiducci 9
review
La diagnosi precoce è fon-
tab. 1 Subset della sclerosi sistemica
damentale per rallentare
l'evoluzione della malattia
SSc Cutanea Diffusa SSc Cutanea Limitata
ed evitare/ridurre i danni
Coinvolgimento Estremità distali e prossimali, Distalmente ai gomiti (acrale) e volto
agli organi interni cutaneo tronco e volto. Presenza di
sfregamenti tendinei da attrito
(tendon friction rubs)
Fenomeno Esordio entro 1 anno dall’inizio Può precedere le manifestazioni
dovute al coinvolgimento polmona- di Raynaud delle alterazioni cutanee (edema cutanee di anni
re, cardiaco (insufficienza cardiaca, o clerosi cutanee) (occasionalmente decadi)
aritmia ventricolare) e renale (crisi re- Coinvolgimento Precoce e significativa incidenza Significativa e tardiva incidenza di
d’organo di fibrosi interstiziale polmonare, ipertensione polmonare, con o senza
nale). L’interessamento polmonare,
insufficienza renale oligurica, malattia interstiziale polmonare,
prima causa di morte si caratterizza malattia gastrointestinale diffusa, nevralgia del trigemino, calcificazioni
in genere per la comparsa di un’in- coinvolgimento miocardico cutanee, cirrosi biliare
terstiziopatia parenchimale associa- Pattern Dilatazioni e rarefazione dei Anse capillari dilatate, di solito senza
capillaroscopico capillari del letto ungueale scomparsa dei capillari
ta o meno ad ipertensione arteriosa
polmonare. Anticorpi Anticorpi anti-topoisomerasi I Alta incidenza di anticorpi
(Scl-70) (30-40%). Assenza anti-centromero (ACA) (70-80%)
degli anticorpi anti-centromero
Diagnosi
Sopravvivenza 2 anni 5 anni
Negli ultimi anni si è comunque as- da inizio malattia
sistito ad una chiara riduzione della
mortalità, con una sopravvivenza a
10 anni dell’80%-90% nei pazienti con tab. 2 Criteri classificativi per la sclerosi sistemica dell’ACR/EULAR (2013)
la forma limitata e circa del 70% nella
forma diffusa, grazie alle diagnosi in Item Sub-Item Score
fasi sempre più precoci di malattia
Fibrosi cutanea estesa prossimalmente 9
e ai numerosi farmaci utilizzati. Una alle articolazioni metacarpofalangee
diagnosi precoce è però fondamen- (criterio sufficiente)
tale per avviare un trattamento che Ispessimento cutaneo delle dita Puffy fingers 2
(considerare solo lo score più alto)
permetta di rallentare l’evoluzione
Sclerodattilia distale alle articolazioni 4
della malattia ed evitare o ridurre i metacarpofalangee ma con
danni agli organi interni, individuan- coinvolgimento della I° falange
do la “window of opportunity” fon- Lesioni ai polpastrelli Ulcere digitali acrali (puntali) 2
damentale a trattare il paziente. (considerare solo lo score più alto)
Cicatrici da vaiolatura sulla punta 3
Segno distintivo della malattia è la delle dita
presenza di autoanticorpi specifici, i Telangectasie 2
principali dei quali infatti sono costitu- Alterazioni capillaroscopiche 2
iti dagli anticorpi anti-topoisomerasi I Interessamento polmonare 2
e dagli anticorpi anti-centromero. Tut- (score massimo pari a 2)
tavia un’alta percentuale dei pazienti Fenomeno di Raynaud 3
SSc è sieronegativo a questi anticorpi, Anticorpi SSc-Specifici (anti-centromero, 3
anti-Topoisomerasi I [ScI70] O anti-RNA
risultando positivo solo alla ricerca di polimerasi III) (score massimo pari a 3)
anticorpi quali anti-Pm-Scl, anti-Th/To,
anti-U3RNP/anti-fibrillarina e anti-RNA
polimerasi I, II e III. in cui l’impegno sclerotico della cute impegno degli organi interni. Fino a
La diagnosi di SSc è piuttosto sempli- coesiste con il FR e con manifestazio- pochi anni fa, la diagnosi di SSc ve-
ce nelle fasi di malattia conclamata, ni o reperti laboristico-strumentali di niva effettuata in base ai criteri ACR
10 MR La Rivista Italiana delle Malattie Rare anno V - n. 1 - febbraio 2021la Rivista
La Rivista Italiana delle Malattie Rare
cercato di identificare criteri che in-
Definizione degli item/sub-item dei criteri classificativi per la
tab. 3 dividuassero tale fase di malattia,
SSc dell’ACR/EULAR (2013)
requisito fondamentale al fine di
prevenire l’evoluzione verso il danno
Ispessimento della pelle Ispessimento o indurimento della pelle non dovuto a cicatrici dopo
lesioni, traumi, ecc. d’organo irreversibile e invalidante.
Dita gonfie Gonfiore delle dita: aumento diffuso, solitamente senza vaiolatura, della In quest’ottica si inserisce lo studio
massa dei tessuti molli delle dita che si estende oltre i normali confini prospettico osservazionale multicen-
della capsula articolare. Le dita normali sono rastremate distalmente
con i tessuti che seguono i contorni dell'osso digitale e delle strutture trico VEDOSS (Very Early Diagnosis
articolari. Il gonfiore delle dita cancella questi contorni. Non dovuto ad Of Systemic Sclerosis) promosso da
altri motivi come la dattilite infiammatoria.
EUSTAR (European Scleroderma Trials
Ulcere della punta delle Ulcere o cicatrici distali o all'articolazione PIP non si ritiene siano dovute And Reasearch Group) e iniziato nel
dita o cicatrici da vaiolatura a traumi. Le cicatrici da vaiolatura digitale sono aree depresse sulle
punte digitali a causa di ischemia, piuttosto che traumi o cause esogene. marzo 2010 al fine di validare criteri
Telangiectasia Le teleangectasie sono vasi sanguigni superficiali dilatati maculari visibili per la diagnosi di SSc in fase molto
che collassano alla pressione e si riempiono lentamente quando la precoce.
pressione viene rilasciata. Le teleangectasie in uno schema simile allo
scleroderma sono tonde e ben delimitate e si trovano su mani, labbra, Come necessari per la diagnosi di
all'interno della bocca e/o grandi teleangectasie opache. Distinguibili da SSc in fase molto precoce sono sta-
angiomi a ragno a riempimento rapido con arteriola centrale e da vasi
superficiali dilatati.
ti identificati quattro segni/sintomi:
presenza di FR, puffy fingers, auto-
Schema capillare Capillari ingranditi e/o perdita capillare con o senza emorragie
della piega ungueale peri-capillari a livello dell'unghia che possono essere visibili sulla anticorpi specifici per SSc (anti-cen-
anormale coerente con SSc cuticola. tromero e anti-topoisomerasi I) e
Ipertensione arteriosa Ipertensione arteriosa polmonare diagnosticata mediante cateterizzazione anomalie capillari compatibili con
polmonare del cuore destro secondo le definizioni standard.
pattern sclerodermico alla capillaro-
Malattia polmonare Fibrosi polmonare alla TC ad alta risoluzione o alla radiografia del torace,
interstiziale più pronunciata nelle porzioni basilari dei polmoni, o presenza di crepitii
scopia. Si tratta di Red Flags che de-
di "velcro" all'auscultazione non dovuti a un'altra causa come l’insuffici- vono far sospettare precocemente
enza cardiaca congestizia. la presenza di SSc, e quindi inviare il
Fenomeno di Raynaud Autovalutazione o segnalazione medica di almeno un cambiamento di paziente ad uno specialista.
colore in due fasi delle dita e spesso dei piedi costituito da pallore,
cianosi e/o iperemia reattiva in risposta all'esposizione al freddo o alle Nel 2013 infine, sono stati creati da
emozioni; di solito una fase è pallore. ACR e da EULAR (European League
Anticorpi specifici Anticorpo anti-centromero o pattern centromero su test di anticorpi Against Rheumatism) nuovi crite-
per la sclerodermia antinucleari (ANA); anticorpo anti-topoisomerasi I (noto anche come
anticorpo anti-Scl70); o anticorpo anti-RNA polimerasi III. Positivo
ri classificativi per la SSc (Tab. 2 e
secondo gli standard di laboratorio locali. Tab. 3), che prevedono la somma
dei punteggi relativi agli item (ispes-
simento cutaneo delle dita, lesioni
La presenza di Red Flags Purtroppo questi criteri consentono fingertip, teleangectasie, anomalie
una diagnosi solo in fasi caratterizza- capillari del letto ungueale, pneu-
deve portare al sospetto
te da fibrosi tissutale, che è solo par- mopatia interstiziale, FR, autoanti-
precoce di SSc e all'invio del
zialmente reversibile e può portare a corpi SSc etc) che risultano positivi.
paziente a uno specialista disfunzioni degli organi colpiti. Le Roy I pazienti con punteggio ≥9 vengono
e Medsger, proposero i criteri per de- classificati come affetti da SSc. Co-
finire la SSc in una fase più precoce, munque, autoanticorpi e pattern
1980 (American College of Rheuma- come una condizione caratterizzata videocapillaroscopico sono consi-
tology) che richiedono la presenza di dal FR, in presenza di anticorpi anti- derati principali strumenti diagnostici
sclerosi cutanea prossimalmente alle nucleari marcatori o di tipiche alte- per indagare i pazienti in presenza di
articolazioni metacarpofalangee razioni capillaroscopiche (megaca- FR, nel sospetto di SSc in fase molto
o metatarsofalangee, o di due dei pillari e/o aree avascolari). Al fine di precoce di malattia.
tre criteri secondari (sclerodattilia, superare però il limite di una diagnosi La videocapillaroscopia periunguea-
ulcere digitali, o fibrosi polmonare). sempre più precoce di malattia,si è le è il metodo più affidabile, non inva-
La sclerosi sistemica S. Guiducci 11sivo, facilmente ripetibile, un registro italiano della
Evoluzione della SSc e finestra di opportunità
per valutare sia la com- fig. 1 patologia volto a de-
per il trattamento
parsa, sia la progressione terminare l’incidenza a
dell’alterazione a livello SSC CONCLAMATA
livello italiano e aperto
SSC MOLTO PRECOCE SSC PRECOCE fibrosi & atrofia
della microcircolazione allo sviluppo di progetti
e quindi per distingue- scientifici.
re precocemente tra FR Puffy fingers fibrosi cutanea & atrofia Il significato che questa
primario e secondario a ANA esofagea/anale patologia rara assume
connettiviti. NVC nella vita del paziente
ACA/ATA cuore, polmone, rene
e della propria fami-
Follow-up Fenomeno di Raynaud ulcere digitali glia, è stata facilitata
I pazienti che presentano dallo studio delle ope-
le Red Flags (FR, edema re dell’eclettico artista
digitale, capillaroscopia alterata, au- Quattro le Red Flags per la Paul Klee, affetto da SSc a metà de-
toanticorpi specifici) dovrebbero es- diagnosi molto precoce: FR, gli anni Trenta del Novecento, ren-
sere sottoposti ad ulteriori indagini per dendo possibile aggiungere tasselli
Puffy fingers, autoanticorpi
valutare la funzione e lo status degli importanti sulla parte emotiva della
organi interni (ecocolordoppler car-
SSc e anomalie capillari malattia, capendone, attraverso le
diaco con misurazione delle pressioni sue opere, la tragedia.
polmonari, TC torace ad alta risoluzio- Il medico deve saper comunicare
ne, prove di funzionalità respiratoria Essendo classificata come malattia con efficacia e con chiarezza con
con misurazione del DLCO, funzionali- rara sono stati istituiti diversi registri i pazienti e con i familiari, sia nella
tà renale delle 24 ore con valutazione regionali che consentono la ge- fase diagnostica, sia in quella del-
della proteinuira e microalbuminuria stione dei dati relativi alle modalità la comunicazione della diagnosi,
ed elettroliti, dosaggio NT-proBNP etc). di presentazione e la prescrizione con particolare riguardo ai pazienti
Ciò richiede una stretta collaborazio- di farmaci; inoltre il registro EUSTAR più giovani che al momento della
ne tra il reumatologo e i medici di me- continua ad arruolare pazienti per diagnosi presentano solo le “mani
dicina generale che devono essere progetti di ricerca scientifica ed epi- fredde” e non sospettano invece, di
pertanto adeguatamente formati a demiologica, infine la SIR (Società avere una patologia che può cam-
riconoscere tali Red Flags (Fig. 1). Italiana di Reumatologia) ha creato biare per sempre la loro vita.
Bibliografia
1. Chiffolt H. Fautrel B, Sordet C, Chateleu E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systemic literature review . Semin Arhritis Rheum. 2008;
37(4) :223-235.
2. Steen VD, Medseger TA. Changes in causes of death in systemic sclerosis,1972-2002.Annals of Rheumatic Diseases 2007;66:940-944.
3. Valentini G, Matucci Cerinic M. Disease specific quality indicators, guidelines and outcome measures in scleroderma. Clin Exp Rheumatol 2007; 25
(47):159-162.
4. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum 2005; 35:35-42.
5. Cutolo M, Pizzorni C, Tuccio M, et al. Nailfold videocapillaroscopic patterns and serum autoantibodies in systemic sclerosis. Rheumatology 2004; 43: 719-26.
6. Hudson M, Thombs B, Baron M. Time to Diagnosis in Systemic Sclerosis: Is Gender a factor? Arthritis Rheum 2007;56:487-489.
7. Guiducci S, Giacomelli R, Matucci Cerinic M. Vascular complications of scleroderma. Autoimmunity Rev 6 2007: 520-523.
8. Matucci Cerinic M, Allanore Y, Czirják L, et al. The challenge of early systemic sclerosis for the EULAR Scleroderma Trial and Research group (EUSTAR) com-
munity. It is time to cut the Gordian knot and develop a prevention or rescue strategy. Ann Rheum Dis. 2009;68(9):1377–1380.
9. Avouac J, Fransen J, Walker UA, et al. Preliminary criteria for the very early diagnosis of systemic sclerosis: results of a Delphi Consensus Study from EULAR
Scleroderma Trials and Research Group. Ann Rheum Dis. 2011;70(3):476–481.
10. van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League
against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737-47.
12 MR La Rivista Italiana delle Malattie Rare anno V - n. 1 - febbraio 2021Angelo Selicorni1, Giuseppe Zampino2,3
l'opinione
1
UOC Pediatria ASST Lariana, Como; 2UOC Pediatria, Fondazione Policlinico Universitario
A Gemelli - IRCCS, Roma; 3Dipartimento di Scienza della Vita e Sanità, Università Cattolica
del Sacro Cuore, Roma
La pediatria della disabilità:
tutti convocati
La letteratura internazionale definisce “bambi- L’impatto di questi pazienti sull’utilizzo dei servizi sani-
ni con bisogni speciali di cura” quei bambini che tari è altrettanto importante se è vero come è vero
hanno o sono a rischio aumentato di essere affetti che questi bambini rappresentano circa l’80% dei
da una condizione cronica su base fisica, evolutiva, pazienti con accessi multipli ai servizi di Emergenza,
comportamentale o emozionale e che richiedono sono responsabili di quasi l’80% dei giorni di utilizzo
prestazioni sanitarie che, per tipologia o quantità, delle degenze delle Rianimazioni pediatriche, quasi
sono superiori a quelle richieste dai bambini della dell’85% dei giorni di utilizzo di ventilazioni assistite in
popolazione generale”. Essi rappresentano circa il Rianimazioni e di più del 50% dei decessi pediatrici.
10-16% della popolazione pediatrica. Al loro inter- Tutti questi dati confermano in modo chiaro l’asso-
no, la stessa letteratura identifica i bambini medi- luta attualità del tema di come organizzare l’assi-
calmente complessi (BMC) che sono “caratterizzati stenza di questi pazienti. Una semplificazione clini-
dalla presenza di problemi sanitari cronici multipli ca permette di differenziare le necessità sanitarie di
che colpiscono diversi organi o apparati; tali pro- questi bambini in 3 grandi aree: necessità del tutto
blemi creano significative limitazioni funzionali, un sovrapponibili a quelle della popolazione pediatri-
frequente utilizzo delle strutture sanitarie ed una fre- ca generale (monitoraggio crescita e sviluppo psi-
quente dipendenza dall’utilizzo di devices”. Questi comotorio, nutrizione, vaccinazioni, intercorrenze cli-
bambini rappresentano circa l’1% della popolazio- niche banali), necessità spesso trasversali ai pazienti
ne pediatrica stessa. È sin troppo ovvio come da con patologie complesse (problematiche degluti-
un lato i pazienti affetti da malattie rare rientrino torie, gastrointestinali, respiratorie, del sonno, della
perfettamente in queste due categorie e come diagnosi e gestione del dolore, gestione routinaria
dall’altro lato questi enormi gruppi coinvolgano dei device) e necessità specialistiche, differenti da
pazienti estremamente eterogenei sul piano eziolo- bambino a bambino e da condizione a condizio-
gico, assistenziale, prognostico e terapeutico. ne (cardiologiche, neurologiche, neurosensoriali or-
L’altro dato unanime derivante dai dati disponibili è topediche, ecc). A fianco delle criticità cliniche si
che la presenza di un figlio affetto da una di queste aggiungono spesso anche quelle riabilitative (mo-
condizioni comporta un impegno assistenziale ed torie, comunicative, cognitive e comportamentali)
organizzativo enorme per gli stessi nuclei familiari. e quelle sociali (inclusione scolastica, gestione del
Secondo Kuo et al. i caregiver di un BMC dedicano tempo libero, supporto al nucleo familiare).
circa 2 ore la settimana per coordinare i vari inter-
venti assistenziali e più di 11 ore la settimana in assi-
Il bambino con patologia complessa dovrebbe
stenza diretta; più della metà delle famiglie hanno
trovare riferimenti significativi, oltre che nel
modificato la loro capacità di lavorare ed il 57% di
centro specialistico di III livello, anche nel
esse segnala la presenza di problemi finanziari. La
pediatra di famiglia e dell'ospedale territoriale
stessa ricerca Diaspro Rosso (2012) ha segnalato
come il 26,8% delle famiglie di una persona affet-
ta da malattia rara fatichi ad arrivare economica- È sin troppo ovvio che un’assistenza così articolata
mente alla fine del mese. e complessa non possa prescindere dal coinvolgi-
La pediatria della disabilità: tutti convocati A. Selicorni, G. Zampino
13mento di multipli attori, molti dei quali strettamente specialistico di III livello, anche nel pediatra di fa-
connessi al territorio di appartenenza della famiglia, miglia e dell'ospedale territoriale.
e tra loro coordinati nel modo più efficiente possibi- Affinché ciò si realizzi è necessario investire in ter-
le. Questo significa che è indispensabile stabilire un mini formativi sulle figure assistenziali territoriali
collegamento virtuoso tra centri di III livello, in gra- che, in primis, dovranno sentirsi parte attiva ed in-
do di garantire il monitoraggio sub specialistico pe- sostituibile del percorso di cura del bambino. Sarà
riodico, la gestione di problematiche complesse in gioco forza importante implementare tutti quei
ambito chirurgico ed internistico, l’impostazione di percorsi informativi e di condivisione dei dati, ine-
percorsi riabilitativi mirati o l’accesso alle eventuali luttabilmente fondati su un utilizzo più diffuso degli
terapie più recenti, e le strutture/figure assistenziali strumenti offerti dal web, nell’ottica di considerare
del territorio (pediatra di famiglia, centro ospeda- la comunicazione tra specialisti e tra diverse figu-
liero provinciale, centro riabilitativo territoriale). Da re assistenziali, come il primo strumento di “buona
questo deriva la necessità assoluta di favorire una pratica clinica”. Ma lo sforzo più importante dovrà
crescita culturale e gestionale capillare in quella essere quello della definizione di un coordinamen-
che è stata definita “pediatria della disabilità” che to delle cure che dia ad uno degli attori il compi-
in termini molto generali potremmo classificare to di svolgere quel ruolo di collante e facilitatore,
come quell’area della pratica clinica che pone at- al tempo stesso, tra le diverse anime del percorso
tenzione e che è in grado di offrire risposte, se non assistenziale. L’obiettivo alto deve essere sollevare
esaustive almeno iniziali, alle problematiche affe- il più possibile la famiglia dalle incombenze orga-
renti alle due prime aree sopra citate. nizzative e garantire che gli interventi messi in atto
derivino da una visione unitaria e condivisa di quel
bambino, di quella famiglia, in quello specifico
La complessità assistenziale di questi
contesto sociale ed emotivo.
pazienti sottolinea l'esistenza
In conclusione, quindi, la complessità assistenziale
di un compito specifico per ciascun attore
di questi pazienti non autorizza alcun attore della
della rete sociosanitaria
rete socio sanitaria a chiamarsi fuori ma, anzi, sot-
tolinea in modo chiaro l’esistenza di un compito
Al fine di offrire al bambino ed alla sua famiglia un specifico e peculiare per ciascuno, per l’adempi-
percorso assistenziale “accessibile, centrato sulla mento del quale tutti sono chiamati di continuo a
famiglia, continuo, coordinato, compassionevole consolidare le proprie conoscenze e competenze.
e rispettoso delle peculiarità culturali”, come de- Parafrasando un celebre monito di Madre Teresa
finito dall'American Academy of Peditrics (AAP) di Calcutta “Quello che ci è chiesto di fare è solo
il bambino con patologia complessa dovrebbe una goccia nell'oceano, ma se non lo facessimo
trovare riferimenti significativi, oltre che nel centro l'oceano avrebbe una goccia in meno”.
Bibliografia
• American Academy of Pediatrics; Medical Home Initiatives for Children With Special Needs Project Advisory Committee. The
medical home Pediatrics.2002;110:184–186.
• Antonelli RC, Stille CJ, Antonelli DM. Care Coordination for Children and Youth With Special Health Care Needs: A Descriptive,
Multisite Study of Activities, Personnel Costs, and Outcomes Pediatrics 2008;122;e209-e216.
• Astolfo R, Bragagnolo P, Porchia S. Progetto Diaspro Rosso 2012 Federazione Italiana Malattie Rare UNIAMO F.I.M.R. onlus.
• Ferreira C. The burden of rare disease Am J Med Genet. 2019;179A:885–892.
• Kuo DZ, Cohen E, Agrawal R, et al.. A national profile of caregiver challenges among more medically complex children with spe-
cial health care needs. Arch Pediatr Adolesc Med. 2011;165(11):1020–1026.
• Kuo DZ, Houtrow AJ, FAAP, Council on children with disabilites. Recognition and Management of Medical Complexity Pediatrics
2016;138(6):e20163021.
• Zampino G, Selicorni A. La pediatria della disabilità. Prospettive in Pediatria 2010; 40 (159-160):89-101.
• Zanello E, Calugi S, Sanders LM, et al. Care coordination for children with special health care needs: a cohort study. Ital J Pedia-
trics 2017;43(1):18.
14 MR La Rivista Italiana delle Malattie Rare anno V - n. 1 - febbraio 2021caso
il Letizia Roggero1, Federico Pieruzzi1,2
1
Dipartimento di Medicina e Chirurgia, Università degli Studi di Milano - Bicocca; 2Clinica Nefrologica
clinico ASST- Monza, Ospedale San Gerardo
Malattia di Fabry con cardiomiopatia
ipertrofica
Riportiamo il caso clinico aneddotico di un uomo all’età di 48 anni, anche il nostro paziente è risulta-
affetto da Malattia di Fabry (MF), variante car- to positivo all’indagine genetica per MF. Il pazien-
diaca, la cui diagnosi effettuata per screening te, successivamente studiato, dal punto di vista
familiare fu inizialmente misconosciuta e classifi- sistemico presentava: un lieve interessamento ne-
cata come cardiomiopatia ipertrofica idiopatica frologico con microalbuminuria, ipoacusia neu-
(Fig.1). La diagnosi di MF, nel contesto delle car- rosensoriale, acufeni e modesti disturbi gastroin-
diomiopatie ipertofiche, è spesso di esclusione testinali con alterazioni dell’alvo. Diversamente,
(Tab.1); poniamo l’attenzione sulla necessità del non presentava acroparestesie, angiocheratomi,
riconoscimento delle manifestazioni sistemiche e cornea verticillata, né coinvolgimento neurologi-
cardiologiche specifiche di questa patologia, che co centrale.
ne permettano la precoce identificazione (1). Nel 2011 fu posto in terapia enzimatica sostituti-
va (ERT) con algasidasi alfa in regime domicilia-
Decorso clinico
Uomo di 60 anni affetto da MF, portatore della
mutazione p.N215S, variante late-onset. Nel 1994, Alterazioni ecocardiografiche
fig. 1
del paziente
all’età di 34 anni, ricovero ospedaliero per polmo-
nite e pericardite, in quell’occasione evidenza
di alterazioni elettrocardiografiche (IVS e onda Finestra di Opportunità
T invertita). Dal 2004 il paziente era in follow-up
cardiologico per il riscontro di cardiomiopatia
ipertrofica familiare, presentava alterazioni eco-
cardiografiche quali ipertrofia ventricolare sinistra
simmetrica (LV massa 161 g/m2) e ipertrofia ventri-
colare del setto (SIV 17 mm) (Fig 1).
Nel 2008 venne posta diagnosi di MF nel fratello,
anche lui in follow-up cardiologico per cardiomio-
patia ipertensiva. In seguito a screening familiare,
La diagnosi di MF è spesso di esclu- A: ecocardiogamma M mode in proiezione parasternale in asse corto,
sione ed il riconoscimento delle sue con evidenza di aumentati spessori parietali del ventricolo sinistro.
B: risonanza magnetica cardiaca con studio del T1 mapping. Valori
ridotti di T1 a livello del miocardio ventricolare sinistro.
manifestazioni sistemiche e cardiolo- C-D: risonanza magnetica cardiaca con delayed enhancement che
evidenza la fibrosi miocardica in sede postero-laterale, severa
giche ne permette la identificazione ipertrofia ventricolare diffusa e significativo aumento della massa
miocardica.
precoce
Malattia di Fabry con cardiomiopatia ipertrofica L. Roggero, F. Pieruzzi
15il caso clinico
La familiarità, X-Linked nella MF, può guidare la
Patologie che possono determinare
tab. 1 ipertrofia ventricolare analogamente diagnosi, così come l’interessamento sistemico
alla MF (3). Raramente, gli esami di imaging permettono
di differenziare le cause di cardiomiopatia con
Cardiomiopatia ipertrofica idiopatica
certezza. La risonanza magnetica cardiaca non
Amiloidosi distingue le diverse forme di cardiopatia ipertrofi-
Sarcoidosi ca sulla base della morfologia ventricolare o degli
Emocromatosi spessori parietali, ma grazie all’utilizzo del mezzo
Malattie da accumulo lisosomiale di contrasto ed alle recenti mappature disponibi-
li, è la tecnica attualmente più idonea per porre
Le caratteristiche di imaging, spesso una corretta diagnosi differenziale. Numerosi studi
hanno, infatti, dimostrato una localizzazione pre-
non dirimenti nella diagnosi differenzia-
ferenziale delle aree di “delayed enhancement”
le delle cause di cardiomiopatia iper- a livello della parete basale infero-laterale e po-
trofica, pongono la necessità di esclu- sterolaterale, presente anche in assenza di fran-
dere diverse patologie sistemiche ca ipertrofia ventricolare. Tale localizzazione può
rappresentare un elemento di sospetto per MF
nell’ambito della diagnosi differenziale in sogget-
re. Con la disponibilità della terapia chaperonica ti con cardiomiopatia ipertrofica idiopatica (4).
orale nel 2018, su richiesta del paziente e per il pro- Inoltre, la risonanza magnetica con mappatura T1
gressivo peggioramento clinico definito sui para- può suggerire un coinvolgimento cardiaco speci-
metri ecocardiografici e sull’instabilità clinica de- fico di MF, in quanto il riscontro di bassi valori di T1,
finita mediante il Fabry Stabilization Index, venne evidenziati solo nei pazienti affetti da MF, riflette-
sostituita la ERT con migalastat. rebbe l’accumulo di sfingolipidi nei miocardiociti,
che sono associati a cambiamenti strutturali ed
Discussione elettrocardiografici precoci (5).
Le caratteristiche di imaging spesso non sono di- Non bisogna trascurare l’importante contributo
rimenti nella diagnosi differenziale delle cause di dell’elettrocardiogramma nella diagnosi differen-
cardiomiopatia ipertrofica e possono porre il cli- ziale delle cardiomiopatie ipertrofiche. Infatti, tra
nico davanti alla necessità di escludere diverse le alterazioni elettrocardiografiche significative,
patologie sistemiche (amiloidosi; malattie da ac- nei pazienti affetti da MF si possono rilevare vol-
cumulo lisosomiale, sarcoidosi, etc) (Tab.1). taggi elevati con marcate anomalie della ripola-
Le caratteristiche ecocardiografiche potevano rizzazione, indipendenti dalla sintomatologia clini-
suggerire la diagnosi di amiloidosi, esclusa, inve- ca e dal grado di IVS. Inoltre, nel 15% dei casi si ha
ce, dagli esami laboratoristici. Diversamente la fa- il riscontro di un PR corto con una conduzione
miliarità ed il coinvolgimento sistemico potevano atrioventricolare che, nelle fasi più avanzate, può
suggerire la MF. evolvere verso blocchi atrio-ventricolari di vario
La cardiomiopatia ipertrofica è un riscontro fre- grado (3).
quente nella pratica clinica; lo studio di questa pa- Il coinvolgimento sistemico può aiutare nella dia-
tologia, dal punto di vista genetico e dell’imaging, gnosi differenziale soprattutto nelle forme classi-
ha permesso di rivelare come in realtà questa de- che di MF, per la presenza di sintomi caratteristici:
finizione comprenda un insieme di patologie mol- acroparestesie soprattutto delle mani e dei piedi,
to eterogenee tra loro, seppur caratterizzate tutte febbri ricorrenti sine causa, disturbi gastrointesti-
dall’IVS. Nella MF l’età di coinvolgimento cardiaco nali, anomalie della sudorazione, ipoacusia neu-
è solitamente più tardiva rispetto alle altre forme di rosensoriale ed acufeni, coinvolgimento nefrologi-
cardiomiopatia, ma più precoce della cardiomio- co ed alterazioni neurologiche quali ictus e
patia amiloidotica da transtiretina (2). attacco ischemico transitorio (TIA). Nel caso clini-
16 MR La Rivista Italiana delle Malattie Rare anno V - n. 1 - febbraio 2021la Rivista
La Rivista Italiana delle Malattie Rare
co riportato, invece, la diagnosi può essere anco- Nelle forme tardive e nelle varianti
ra più insidiosa dato che nelle forme tardive o nel-
le varianti cardiache di MF (come per la variante
cardiache di MF la cardiomiopatia
genica p.N215S), la cardiomiopatia può essere può essere l'unica manifestazione cli-
l’unica manifestazione clinica significativa. nica significativa
Conclusioni diagnostica differenziale e soprattutto un accu-
Appare evidente come la MF possa simulare la rato e specifico utilizzo degli esami di imaging, in
diagnosi di diverse forme di cardiomiopatia iper- quanto sono disponibili terapie specifiche in gra-
trofica, inducendo a diagnosi errate o molto ri- do, se avviate precocemente, di avere un impatto
tardate. Risultano di fondamentale importanza la favorevole sulla prognosi.
Bibliografia
1. Ommen SR, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A
Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J
Am Coll Cardiol. 2020 Nov 20:S0735- 1097(20)36413-5.
2. Maurer MS, et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ
Heart Fail. 2019;12(9):e006075.
3. Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart. 2007; 93:528-35.
4. De Cobelli F, Esposito A, Belloni E, et al. Delayed-enhanced cardiac MRI for differentiation of Fabry’s disease from symmetric
hypertrophic cardiomyopathy. AJR Am J Roentgenol 2009;192:97- 102.
5. Camporeale A, Pieroni M, Pieruzzi F, et al. Predictors of Clinical Evolution in Prehypertrophic Fabry Disease. Circulation. Cardiova-
scular Imaging, 12(4), e008424.
Una proposta educazionale a 360°:
Un
la Rivista
la Rivista
il portale Web
il Corso online di
Formazione a Distanza
Ogni numero della Rivista è collegato ad una FAD
(Formazione a Distanza), con erogazione dei crediti
formativi ECM
La Rivista Italiana delle Malattie Rare
w w w. m a l a t t i er ar e .eu
per leggere MR e i supplementi,
MR La Rivista Italiana delle Malattie Rare anno V - n. 1 - febbraio 2021 17
richiedere la Rivista, contattare la redazione
e per accedere alla FAD di MRPuoi anche leggere

















































