MAPPANDO L'IGNOTO NELLA PENOMBRA: PROFILI DI BIODIVERSITÀ IN CAMPIONI AMBIENTALI - Saverio Vicario CNR-Istituto Tecnologie Biomediche
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù

MAPPANDO L’IGNOTO NELLA PENOMBRA: PROFILI DI BIODIVERSITÀ IN CAMPIONI AMBIENTALI Saverio Vicario CNR- Istituto Tecnologie Biomediche
Sommario
L’approccio barcode
Profili di biodiversità da sequenziamenti ambientali
Produzione del dato ed errori
Pre-Processamento o pulizia del dato
Proposta di approccio basato su diversità
filogenetica
Risultati di simulazioni e di un caso studio
Barcode
Definizione:
“locus standardizzato per la diagnosi di specie”
Generalizzazione per i profili di biodiversità
“locus/i standardizzati per descrivere la biodiversità”
Il mio punto di vista sul barcode
Quadro teorico
Problemi di concordanza tra concetto filogenetico di
specie e concetto morfo-ecologico
Problemi di concordanza albero di gene e specie
Caso speciale: pseudogeni nucleari dei geni mitocondriali
(numts)
Implementazione
Problemi assegnazione a classe
Correttezza dei metodi vs robustezza e velocità
Quantità e qualità dei campioni di riferimento
Come riconoscere i campioni che non hanno riferimenti validi
Concordanza tra concetti di specie
Osservazione di base al concetto di specie: esiste
una discontinuità nello spazio fenotipico
Concetto morfo-ecologico: la specie sono quel
gruppo di individui con simili attributi in quanto
occupano lo stesso picco adattivo e la selezione
purificante elimina individui divergenti.
Concetto filogenetico: la specie e’ quel gruppo di
individui che attributi simili in quanto appartengono
allo stesso lignaggio e li hanno ereditati da un
ancestore comune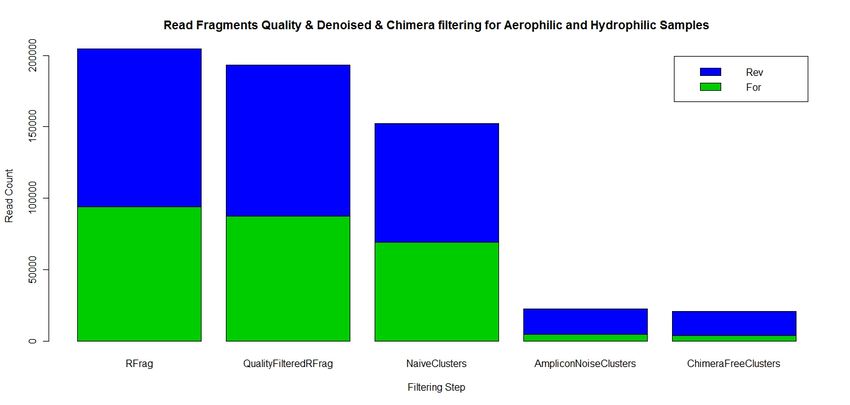
Basi teoriche di un buon riferimento
Specie A Specie B
MRCA
Sulla base della teoria della coalescenza, pochi campioni ( 5-10) se ben distribuiti nelle
popolazioni delle specie possono rapprensetare correttamentela variabilita di un singolo
locus
Sempre per gli eventi di coalescenza ci aspettiamo un aumento della concordanza tra
albero albero di gene e di specie nel caso di marcatori organellari
Infatti, la ridotta dimensione di popolazione del mitocondriale dovrebbe garantire veloce
fissazione e piu rapida coalescenza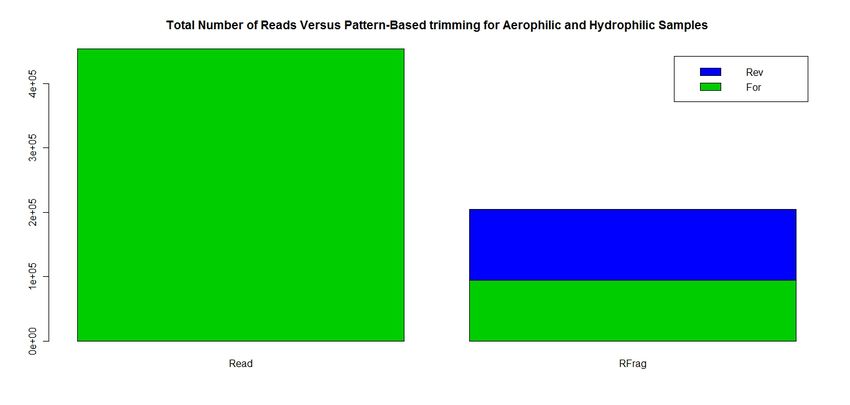
Difficolta nell’assegnazione di specie
Species A
Species B
X
MRCA
Il paradigma cade in caso di una forte struttura di popolazione e di un
campionamento concentrato.
Il rischio di specie con popolazione fortemente strutturate credo sia più
alto in specie temperate dato la più bassa dimensionalità di nicchia
(dominata da fattori abiotici) non facilita l’individuazione di nuovi picchi
adattavi anche in presenza di barriere geografiche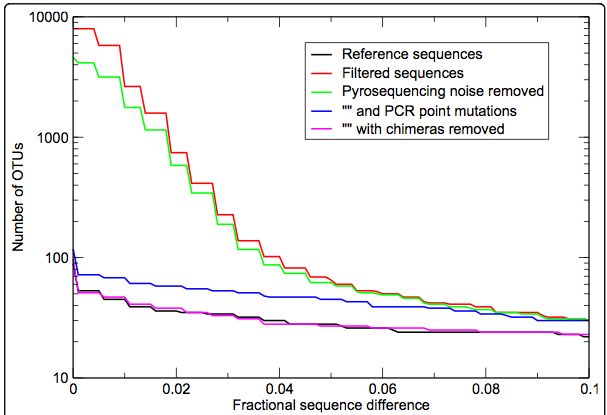
Le distanze confondono
Le distanze non fanno differenza tra i vari siti
La divergenza intraspecifica e’ funzione della struttura
di popolazione, dunque la mancanza di gap e’ solo
indice di un diverso grado di struttura di popolazione,
ma i nodi di ogni singola specie potrebbero essere ben
supportati
K2P andrebbe sostituito almeno con HKY che permette
frequenza basi diverse con un costo computazionale
identico (genoma mitocondriale caratterizzato da forti
squilibri composizionali e moderato rapporto
transizioni /trasversioni)Alberi o non alberi
Preferisco alberi poco definiti a profili di distanze
Sono d’accordo che gli alberi vanno presi
seriamente e credo sia utile abbandonare
neighbour joining e K2p.
FastML, RAXML sono programmi robusti e veloci per
avere approssimazioni di alberi filogenetici di
massima verosimiglianza anche grandi (vedi
progetto SILVA)
L’importante e’ riportare delle stime di supporto
nodaleI profili di biodiversità
Terminologia: un punto di vista
Sequenziamenti Ambientali
sequenziamenti da estratti di DNA/RNA da campioni non isolati
Metagenomica Applicabile
sequenziamenti ambientali con copertura tale da poter ricostruire almeno solo a Batteri,
parzialmente i genomi presenti Archibatteri o
Si ricava principalmente informazione funzionale ad organismi
con genomi
Metagenetica/profilo di biodiversità piccoli
sequenziamenti ambientali mirati su un sottoinsieme di loci
Si ricava informazione filogenetica/tassonomica Usato per affrontare
comunità di Eucarioti e/o
annotare
tassonomicamente
comunità BattericheProfili di biodiversità
I sequenziamenti ambientali con lo scopo di
esplorare i profili tassonomici producono alla fine
una lista di sequenze con un attributo di
abbondanza.
Queste sequenze possono essere poi raggruppate
in OTU o MOTU (cluster con soglia arbitraria di
divergenza, parola con diverso uso che in contesto
sistematico) o specie o altri tipi di categorie
biologicamente rilevanti
Lo scopo e’ generalmente quello di distinguere i
profili di abbondanze tra due o più ambienti diversiFormalizzazione
Categorie: Ambiente I Ambiente II
Sequenza
unica/OTU/
specie
Replicato I Replicato II Replicato I Replicato II
1 CI,I,1 CI,II,1 CII,I,1 CII,II,1
2 CI,I,2 CI,II,2 CII,I,2 CII,II,2
… … … … …
n CI,I,n CI,I,n CII,I,n CII,II,n
La domanda e’ dunque se esiste un differenza tra i due
ambienti tenendo conto della variabilità intra campione
Si usano approcci come:
T test con correzioni multipla
Distribuzione di poisson multiple
Distribuzioni multinomialiProduzione del dato
Sequenziamenti di tipo Next Generation Sequences
454 life science read da 400bp (1000bp in prospettiva)
per circa 600mila reads
Ilumina read da 100bp ma con paired end 200-250bp per
decine di millioni di readsErrori nella produzione del dato
Sistematici Fasi di lavoro Soluzioni
Preferenzialità nella Campionamento
Campionamento stratificato
raccolta dei campioni
Difficoltà di estrarre DNA da Preparazione del campione
alcuni organismi ( rivestimenti Estrazione DNA (es. usare solo zampe)
chitinosi, depositi grassi)
Preferenzialita dei primer sia Usare diversi primer per la
universali che random ( bias Amplificazione stessa regione e unire i
composizionale); risultati
strutture secondarie
Strutture secondarie, dimensioni
Libreria …
dei frammenti
Errori di lettura negli
omopolimeri: più errori in Sequenziamento Denoising: es. Ampliconoise
organismi con mtDNA con bias
composizionaleErrori nella produzione del dato
Stocastici Fasi di lavoro Soluzioni
Campionamento Replicati biologici
Campionamento
Inibitori che diminuiscono lo
sforzo campionamento Estrazione DNA Diverse estrazioni da unire
effettivo
Errori di polimerasi Amplificazione Polimerasi ad alta fedelta e Denoising
Campionamento Libreria …
Errori di lettura Sequenziamento DenoisingCampionamento: l’inutilità dei conteggi
La stima dello sforzo di campionamento e dell’errore
non può essere dedotto dal numero dei read
Infatti il numero totale di read e’ totalmente
indipendente dal numero di molecole di DNA
campionate a causa dei molteplici passaggi di
campionamento e ri-amplificazione
I conteggi sono quindi stimatori delle frequenze relative
non assolute
I replicati biologici diventano l’unica maniera per
verificare la varianza di campionamentoSoil Carots PitTraps
OR
Sample processing
Workflow
DNA
Extraction Pyrosequencing
454 Shotgun
PCR with Amplicon Phi29
Nebulization Library
Folmer’s Ligations amplification
Rapid protocol
F R
+Adaptors
670-700bpPrimer bias in dettaglio I
Stime qPCR fatte su 6 repliche indipendenti Usando solo read
con perfect matchPrimer bias in dettaglio II
Usando solo read
Stime qPCR fatte su 6 repliche indipendenti con perfect matchPre-Processamento
Preprocessamento: Denoising
I singoli read NGS hanno una qualità minore dei
read Sanger.
Per descrivere un genoma tipicamente vengono
mappati su una sequenza di riferimento e viene poi
fatta un statistica per ogni sito
Nel caso del sequenziamento ambientale non si sa
quanti genomi/alleli diversi sono presenti
Diverse procedure di clustering per ricostruire il numero
di genomi diversi presente e le sequenze originali
presenti nel DNA estratto totalePreProcessamento:
Esempio da AmpliconNoise
Numero di cluster
Soglia di clustering
Quince et al. BMC Bioinformatics 2011, 12:38Analisi di profili di biodiversità
Analisi del le Categorie I
Le sequenze uniche non hanno attributi informativi
supplementari, inoltre possono variare da un campione
all’altro per ragioni non biologicamente rilevanti ma
contengono tutta l’informazione sulla variabilità del
campione
Le OTU (cluster con soglia fissa) risolvono riducono la
ridondanza dei dati anche se in maniera arbitraria: non
e’ detto che il livello di organizzazione rilevante sia
quello dell’OTU e le OTU essendo arbitrarie non
consentono di usare attributi raccolti in precedenti studiProblemi: le Categorie II
Le specie sono categorie con:
ricca dote di attributi da precedenti studi
Sono spesso, ma non sempre (patogenicità in Fusarium),
biologicamente rilevanti
Non si possono assegnare senza un campione di riferimento
Categorie tassonomiche superiori:
Alcuni attributi utili disponibili
Non sempre biologicamente rilevanti ( anche se vedi es.
flora intestinale)
Necessitano riferimento ma più facile reperire ed assegnare
(es. PhyloPhithia)Proposta di approccio basato su diversità filogenetica
Una soluzione per le categorie: usarle
solo se necessario
Abbandonare il paradigma delle catergorie e
usare un indice di diversità basato sulla struttura
delle relazioni filogenetiche fra le sequenze
Utile per situazioni con poche informazioni
accessorie e comunque pochi attributi disponibili a
qualunque livello tassonomico ( grazie all’albero)
Il problema da multidimensionale diventa
unidimensionale ( grazie all’indice di diversità)Indici di Diversità
Esistono una grande varietà di indici
Si distinguono per tre aspetti:
Peso dato alle diverse frequenze di osservazione
Considerare le categorie come tutte uguali o in un
contesto filogenetico
Additività degli effetti (Al raddoppio del numero di
specie con uguale frequenza corrisponde un aumento
proporzionale dell’indice)Indici di Diversità: diverso uso frequenze
La serie dei numeri di Hill permette di generare una
classe di indici con variabile sensibilità alle basse
frequenze
1/(1−q )
S
q ≠1 q
D = ∑ pi q
i=1
S Non E’ altro che
q = 1 1D = exp −∑ pi log( pi ) l’esponenziale dell’indice di
i=1 Shannon
Valori più bassi di uno favoriscono le basse frequenze
fino ad arrivare a q=0 che e’ il semplice conteggio
delle specie, mentre valori >1 sono più sensibili alle
alte frequenzeDifferenza fra D e H
L’entropia e’ spesso usata per stimare la diversità di un campione, ma non aumenta in
maniera proporzionale al numero degli oggetti
S
q ≠1 q
H = ∑ piq q
D= q H1/(1−q )
i=1
S
q = 1 H = −∑ pi log( pi ) D = exp(H)
Dunque bisogna applicare l’esponenziale per q=1 o l’esponente
i=1 della formula di Hill
Es. per q=2 H e’ uguale alle stime di eterozigosità della genetica di popolazione
mentre D e’ usato per stimare il numero effettivo di codoni per un singolo
aminoacido
H=stima la sorpresa media € di osservatore di fronte ad un datum della serie
D= stima il numero di elementi equiprobabili equivalenti alla diversità del sistema in
osservazione
Gli indici H si sommano per ottenere H totale mentre le D si moltiplicanoRelazione tra la serie di hillis e varie indici
in uso in ecologia
Lou Jost OIKOS 113:2 (2006)
X= indice p= frequenza relativaRelazione tra diversità beta e indici
presi da teoria dell’informazione
Se l’esponenziale dell’entropia di shannon e’ una
buona misura della diversità biologica allora
l’esponenziale della mutua informazione tra la lista
di specie di due ambienti e’ la corretta stima della
diversità beta dei due ambienti
Mutua informazione può essere calcolata come la
media della distanza di Kullback-Leibler tra ogni
ambiente e la media degli ambienti
(Ludovisi, A. et Taticchi, M. I. (2006). Ecological
Modelling 192(1-2): 299-313.)Formalizzazione
S oggetti in C campioni (4) in A ambienti (2)
La diversità Alpha e’ la diversità intra campione
La diversità Beta e’ la diversità inter campione
La diversità gamma e’ la diversità totale
Si assume che :
Dgamma = Dbeta ⋅ Dalpha
H gamma = H beta + H alpha
D Gamma e D alpha sono misurate in numero di
specie equivalenti mentre D Beta e’ numero di
campioni equivalenti
I cerchi sono i campioni, i colori gli Dbeta varia tra 1 e C. Se 1 non c’e’ differenza tra i
ambienti € campioni
Numero di specie equivalenti
condivise tra i campioni ed ambientiCome rappresentare i dati
ID seq Campio Ambien
ne te
1 2 1
1 2 1 Ogni rigo un read (un osservazione)
1 2 1 S= vettore di appartenza a cluster di
2 1 1 denoising
2 3 2 C= vettore di appartenenza a campione
2 3 2 A= vettore di appartenenza ad ambiente
2 3 2
2 3 1
2 3 1
… … …Formalizzazione:
Quantità di informazione
Halpha= H(S|A,C)=H(S|C)= rumore biologico
HbetaC|A= MI(S,C|A)= rumore sperimentale
HbetaA= MI(S,A)=segnale, mutua informazione dell’ambiente e delle sequenze
C
+ + =H(S) S A
Halpha HbetaC|A HbetaA
HbetaC|A
Halpha HbetaA
Se HbetaA =0 allora DbetaA =1
DbetaA numero di ambienti diversi equivalentiFormalizzazione: Come permutare
ID seq Campio Ambien
ne te
1 2 1
1 2 1
1 2 1
2 1 1
2 3 2
2 3 2 Permutazione di ID seq tenendo fermi
gli altri
2 3 2
2 3 1
2 3 1
… … …Indici con informazione filogenetica
In casi di mancanza di dati di riferimento o per
difficoltà di assegnazione oppure perche non si e’
sicuri del livello di diversità che conta tra i campioni
si deve prendere in considerazione la relazione fra
le sequenze uniche
Indici filogenetici:
Senza abbondanze(es. PD di Faith)
Con abbondanze (entropia filogenetica D(T) di chao,
ma anche Allen e Rao)Indici filogenetici con abbondanze
Gli indici filogenetici non usano più come unità il numero
di specie equivalenti ma il numero di lignaggi
equivalenti in una data finestra di tempo
Entropia filogenetica di Allen
n
H p = ∑ Li p j log( p j )
i=1
In cui L e’ la lunghezza di un ramo e p la somma delle
frequenze delle specie che discendono dal quel ramo
Hp non e’ una corretta generalizzazione perché la
€ somma di tutti gli Lp non fa 1Come si calcolano le abbondanze per
ogni ramo
p1
p1+p2
p2
p4+p3+p5
p3
p4+p3
p4
p5Indice di Chao
Chao normalizza Hp per la somma di tutti i rami
pesati per l’abbondanza delle specie discendenti
consentendo di avere una misura che non dipende
dalla sezione di tempo presa in considerazione e
mantiene le caratteristiche di additività degli indici
di diversità normali
2S−1
Li
q = 1 H p = ∑ pi log( pi ) 2S−1
T
i=1
2S−1 T = ∑ Li pi
Li q
q ≠1 q
H p = ∑ pi i=1
i=1 T
Anne Chao, et al. 2010 Phil. Trans. R. Soc. B,365:3599-3609Guardando il dettaglio:
un Hbeta per ogni ramo usando DKL
Usando il valore medio della distanza
di Kullback-Leibler (DKL) della
distribuzione totale di ogni singolo
gruppo si può ottenere una stima di
Hbeta per ogni ramo della filogenesi
Come per il valore beta totale si puo
effettuare una permutazione dei dati e
verificare la significatività di ogni
singolo ramo.
Si pone il problema di scegliere quali
rami prendere in considerazione visto
che alcuni sono un sottoinsieme di altri.
Credo che non ci sia problema a
generare le diverse distribuzioni per
ogni ramo usando la stessa
permutazione
Più problematico e’ il problema del test
multipli.Problemi di stima di entropia
L’entropia e’ molto sensibile ad errori nella stima di
basse frequenze, per questo in ambito ecologico e’
stato proposto la correzione all’indice di Shannon
Si assume che specie non osservate
n1 siano quante quelle osservate una
C = 1−
n volta
s
Cpi log(Cpi )
HChaoShen = ∑ n
i=1 1− (1− Cpi )
Non applicabile all’entropia filogenetica
Anne Chao et al . 2003. Environmental and Ecological Statistics 10, 429-443
€Problemi stima indici entropia
filogenetica
L’entropia filogenetica e’ più robusta di un semplice
Shannon se gli eventi rari non campionati sono
varianti simili delle categorie già trovate. Infatti
questi eventi verranno pesati poco essendo connessi
all’albero con rami corti.
L’entropia filogenetica e’ invece molto sensibile ad
una categoria rara molto divergente. Le curve di
rarefazione della divergenza dei cluster possono
aiutare a capire se le differenze riscontrate tra due
ambienti sono imputabili solo a loro.Come implementare approcci
computazionalmente costosi
Un approccio basato sulla diversità filogenetica per
un progetto di sequenziamento ambientale
necessita di avere una stima di albero filogenetico
di circa 100-20mila taxon.
Possibile spezzare il problema in gruppi di qualche
migliaio di taxon con categorizzazioni a grossi
gruppi tassonomici usando altri tipi di classificazioni
Affrontare il problema di petto con un ML
approssimato (RaXML ligth )Ma quale albero filogenetico?
COI o altri marker non hanno risoluzione a media o
profonda divergenza
Possibile però usare constraint topologiche prese
progetti Tree of Lifeo comunque dalla tassonomia di
riferimento
Usare modelli dettagliati per estrarre tutta
l’informazione filogenetica possibileCome far crescere le comunità
Creare delle infrastrutture per la comunita dello studio
della biodiverista
Accesso alle banche date
Tassonomiche
Osservazioni
Molecolari
Accesso a potere di calcolo
Accesso a programmi, che segua le innovazione della
comunita’
Accesso a workflow che includono le best practice
migliorabili dalla comunita degli utenti Infrastruttura ESFRI (www.lifewatch.eu), al momento solo
su carta, anche se esiste una forma embrionale di
governance (www.lifewatch.it) e il MIUR ha riconosciuto
l’iniziativa.
Giornata Nazionale di Presentazione dell’infrastruttura
LifeWatch
Roma 14 dicembre 2011 ore 14:30
Palazzetto Mattei, Villa Celimontana, c/o Società
Geografica Italiana
via della Navicella 12, Roma. Primi progetti di costruzione con coinvolgimento
italiano
Biovel [www.biovel.eu](FP7) per lo sviluppo di
webservices di interesse per la biodiversità e di
possibili workflow (argomenti iniziali: accesso a banche
date tassonomiche, inferenza filogenetica [raxml,TNT,
mrbayes], stima nicchia potenziale [openmodeller],
modelli per fissazione della CO2 [BIOME-BGC])
PON strutturale Bio4IU INFRASTRUTTURA MULTIDISCIPLINARE PER
LO STUDIO E LA VALORIZZAZIONE DELLA BIODIVERSITÀ MARINA E
TERRESTRE NELLA PROSPETTIVA DELLA INNOVATION UNIONBio4IU L’infrastruttura Bio4IU è finalizzata allo studio degli organismi viventi e dei meccanismi alla base del mantenimento della biodiversità. Il progetto prevede la definizione e l’attivazione di interventi di adeguamento e rafforzamento strutturale riferiti a cinque nodi, alcuni dei quali costituiti dalla convergenza di più Istituti.
Bio4IU BIOforIU è articolata in componenti tematiche, con un nodo principale e altri nodi partecipanti, connesse alle diverse infrastrutture ESFRI.
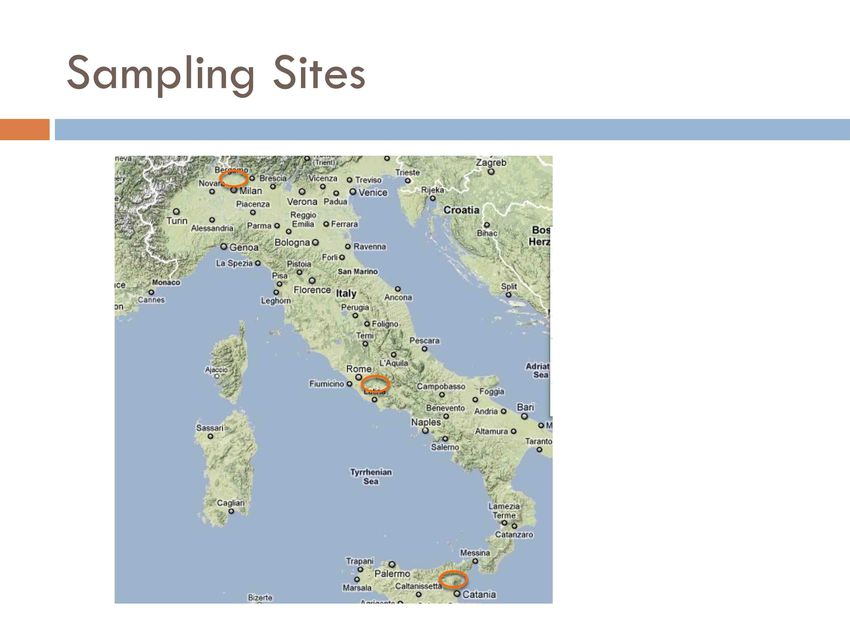
Example of implementation Metazoan diversity of chestnut soil forest Objective: Exploring efficiency of NGS sequencing in giving a complete profile of soil metazoan diversity Sampling: Chestnut forest soil was selected because is the less biodiverse forest soil in Italy. In fact, Chestnut forest are not italian native but are an effect of Ancient Roman introduction. No real chestnut italian species specialist exist. This average complexity was preferred at the level of experiment design
Sample Summary
Name Locarion Sampling method
NA North Aereobiont
NI North Idrobiont
CA Centre Aereobiont
CI Centre Idrobiont
SA South Aereobiont
SA South Idrobiont
MPE4 Centre Pit Trap
MPE5 Centre Pit Trap
Aereobiont sampled with Berlese funnel from soil of at
least 4 soil carots
Idrobiont sampled with Baerman funnel from soil of at
least 4 soil carotsSoil Carots PitTraps
OR
Sample processing
Workflow
DNA
Extraction Pyrosequencing
454 Shotgun
PCR with Amplicon Phi29
Nebulization Library
Folmer’s Ligations amplification
Rapid protocol
F R
+Adaptors
670-700bpNGS output: quality scores and read numbers
NGS output: read lengths
Section
Section 81
10000
8000 5000
Filtered
Filtered
Unfiltered
Unfiltered
6000 4000
of read
read
4000 3000
N of
2000 2000
N
1000
0
0
50
50 150
150 250
250 350
350 450
450 550
550 650
650 750
750 850
850 950
950 1050
1050 1150
1150
read
readlength
lengthPost NGS in silico Workflow
Primer pattern COI 5’/3’
recognition recognition Denoising Chimera filtering
Global Alignment Blastn+BOLD AmpliconNoise UChime
3’Fragment
3’ Fragments Clusters
Reads
5’ Fragments 5’Fragment
Clusters
3’ 5’ 3’ 5’
100bp Flow signal at
filter least 0.7Post NGS Results:
Primer Pattern and 5’/3’ Recognition
Reads Reads
FragmentsPost NGS Results: Denoising yield
5’
3’
Read Quality Identical AmpliconNoise ChimeraFree
Fragments Filtered Fragment Fragment Fragment
Clusters Clusters ClustersBiodiversity Profiling Workflow
Getting References Align References Align Clusters Infer Phylogenetic Trees
BlastX/HMMer3.0 Muscle Hmmer RAxML
Nucleotide
Protein Space Ref. +
Space
Fragment Ref. Order Fragment Ref. + Fragment Clusters
Clusters MSA Clusters Order Order Tree
MSA
Web Services Phylogenetic
Diversity
BOLD DB Local DB Python Script
Lineage that
EmblCDS
differentiateOrder Content of Pit Trap Sample in Log scale
!"
!"
Morphologically observed
!"
Other Insecta
Bacteria !"
Marine orders (Error) !"
Fungi
2% of identified biomass is lost
( Miriapoda)
11% of identified biomass have low scores
(Orthoptera, Scorpions and Spiders)Order in Pit Trap: weight vs. reads
Coleoptera
Hymenoptera
Diptera
Isopoda
10000
Lepidoptera
Blattodea
reads
Collembola
100
Aranae Orthoptera
14% of identified
biomass
Scorpiones
1
0.01 0.05 0.50 5.00
weightEsempio di cambiamento trovato
Nei campioni di Idrobionti nei
read assegnati ai Rhabditida
abbiamo trovato un unico
lignaggio differenziato tra le 3
località
L’intero lignaggio non ha un
neanche una sequenza di
riferimento. Il riferimento piu vicino
e’ un campione giapponese
Ordine Rhabditida
Dbeta =1.24 numero campioni
effettivo
Max(Dbeta)=3 campioni
Specie di riferimento piu’
prossima in giapponeRingraziamenti sul caso studio
CNR-ITB, Bari Università Milano “Bicocca”
Mol. Lab Maurizio Casiraghi
Apollonia Tullo Michela Barbuto
Anna Maria Paluscio Andrea Galimberti
Caterina Manzari Università Roma “TorVergata”
In silico Donatella Cesaroni
Cecilia Lanave Emiliano Trucchi
Giorgio Grillo Angela Fortunato
Flavio Liciulli*
Università di Catania INFN
Università di Bari Bianca Lombardo Giacinto DonVito
Bachir Balech Gipo Montesanto
Funds:
Dedicato a Cecilia Lanave PRIN (MIUR)Fine
Puoi anche leggere

















































