CORSO di CHIMICA (06AHM) - Facoltà di Ingegneria - POLITECNICO di TORINO - Politecnico di ...
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù

Corso di Chimica – A.A. 2014-2015
Facoltà di Ingegneria – POLITECNICO di TORINO
Anno Accademico 2014-2015
CORSO di CHIMICA (06AHM)
ITt
Magritte
Emma Angelini
Dipartimento di Scienza Applicata e Tecnologia - Politecnico di Torino
e-mail : emma.angelini@polito.it
E. Angelini – DISAT – Politecnico di Torino -1-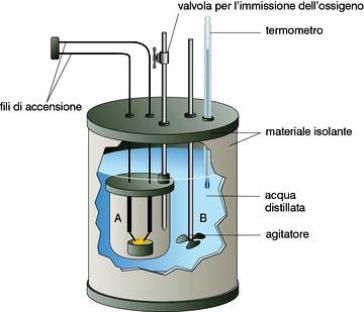
Corso di Chimica – A.A. 2014-2015
LEZIONE 9
TERMODINAMICA: STUDIO DELLE TRASFORMAZIONI DELL'ENERGIA
La termodinamica è la disciplina che descrive le proprietà dei sistemi chimico-fisici dal punto di
vista macroscopico e ne studia il comportamento; in particolare si occupa dei trasferimenti di
energia che si producono nel corso delle trasformazioni e delle modalità con cui queste
avvengono.
L'approccio termodinamico ha il vantaggio di essere indipendente da qualsiasi ipotesi sulla
struttura della materia.
Al fine di comprendere meglio i concetti riguardanti la termodinamica è necessario dare alcune
definizioni.
Sistema: Porzione delimitata dell'universo, alla quale ci riferiamo per le nostre osservazioni. In
particolare I sistemi chimici sono costituiti dai materiali (reagenti e prodotti) che partecipano alle
trasformazioni fisiche e chimiche della materia. Dal punto di vista termodinamico è utile
distinguere tre diversi tipi di sistema in base ai suoi rapporti con l'ambiente.
Ambiente: Tutto ciò che circonda il sistema (è dall'ambiente che si effettuano osservazioni sul
sistema)
Sistema + Ambiente = Universo
Si distinguono tre tipi di sistemi, in base ai rapporti con l'ambiente:
• Sistema aperto - Può scambiare con l'ambiente Materia e Energia
• Sistema chiuso - Può scambiare con l'ambiente solo Energia
• Sistema isolato - Non può effettuare nessun tipo di scambio
I sistemi chiusi possono essere di due tipi: diatermici e adiabatici. I primi possono scambiare con
l'ambiente lavoro e calore, i secondi sono termicamente isolati e possono scambiare lavoro con
l'ambiente, ma non calore.
Per es., un sistema può consistere in una sostanza o in una miscela di sostanze coinvolte in una
reazione chimica o in un passaggio di stato.
Di solito, per ovvi motivi, si considera come ambiente esterno solo la zona situata nelle vicinanze
del sistema. I confini di un sistema possono essere reali, come le pareti di un recipiente, oppure
immaginari, come le superfici di separazione fra le zone dell'atmosfera.
LO STATO DI UN SISTEMA È DEFINITO DALLE SUE PROPRIETÀ
Si distinguono:
• Proprietà intensive - Indipendenti dalle dimensioni del sistema: esempio, P e T
• Proprietà estensive - Dipendenti dalle dimensioni del sistema: esempio, V, massa e funzioni
termodinamiche tipiche.
Talvolta le proprietà estensive vengono espresse in termini relativi (ad esempio, densità =
massa/volume; concentrazione = moli/volume). In tal caso, divengono proprietà intensive e si
definiscono proprietà specifiche, nel senso che caratterizzano una sostanza rispetto alle altre.
E. Angelini – DISAT – Politecnico di Torino -2-Corso di Chimica – A.A. 2014-2015 Le Proprietà di un sistema si definiscono funzioni di stato e sono correlate fra loro da equazioni di stato (ne è un esempio l'equazione generale di stato dei gas). Le funzioni di stato hanno la caratteristica di essere indipendenti dal "percorso" attraverso il quale un determinato stato è stato raggiunto. Ciò significa che le loro variazioni sono esattamente definite dalla differenza fra il valore che hanno nello stato finale e quello che hanno nello stato iniziale. Ad esempio, consideriamo la reazione di ossido-riduzione del glucosio: C6 H12 O6 + 6 O2 à 6 CO2 + 6 H2 O + 2879 kJoule La quantità di energia prodotta è la stessa sia nella combustione semplice che nella respirazione cellulare, l'unica cosa che cambia è il modo secondo cui procede la reazione e viene liberata l'energia: istantaneamente nel primo caso, attraverso una serie di reazioni intermedie catalizzate ciascuna da enzimi e coenzimi nel secondo. I PRINCIPI IN SINTESI I principi della termodinamica sono riconducibili a tre osservazioni sperimentali fondamentali. Ciascuno diessi porta a definire una funzione di stato fondamentale , nell'ordine: Temperatura (T), Energia Interna (U), Entropia (S). 0) "Se due corpi, A e B, sono entrambi in equilibrio termico con un terzo corpo, C, essi sono in equilibrio termico anche fra loro." Questo enunciato è noto come zeresimo principio della termodinamica e sta alla base del concetto di temperatura e della sua misurazione mediante il termometro. 1) "L'energia dell'universo è costante." È una delle possibili formulazioni del primo principio della termodinamica . Non è altro che l'espressione del concetto di conservazione dell'energia (Mayer, 1842). Si introduce con esso la funzione energia interna di un sistema. Consente di stabilire quali trasformazioni siano possibili. 2) "È impossibile invertire completamente un qualsiasi processo naturale." È questa una delle possibili formulazioni del secondo principio. Esso pone delle limitazioni alle trasformazioni di calore in lavoro (Carnot, 1824) e introduce la funzione entropia. Consente di stabilire quali delle trasformazioni possibili siano spontanee. IL PRIMO PRINCIPIO I concetti fondamentali che si incontrano nella formulazione del 1° Principio sono Energia, Lavoro e Calore. Energia: L'energia di un sistema è espressione della sua capacità di compiere Lavoro (ad es., in chimica l'energia del sistema è la somma delle energie cinetica e potenziale delle particelle costituenti.) Lavoro: Dal punto di vista meccanico si definisce come lo scalare F·s (forza per spostamento) e può pertanto essere sempre ricondotto al caso in cui un corpo viene spostato contro una forza. Calore: Un modo alternativo per scambiare energia, diverso dal Lavoro. Quando due corpi a E. Angelini – DISAT – Politecnico di Torino -3-
Corso di Chimica – A.A. 2014-2015 differente T sono posti in contatto termico, vi è un trasferimento di energia, un flusso di calore, da quello a T più elevate a quello a T più bassa. Durante una trasformazione che dai reagenti porta ai prodotti tutte le particelle del sistema si sono riarrangiate spezzando legami e formandone di nuovi per dare i prodotti. Analogamente all’acqua che, finendo a valle, trasforma energia potenziale in energia cinetica, anche le particelle nella trasformazione da reagenti a prodotti hanno sviluppato energia, che chiamiamo Energia interna (U), che si manifesta macroscopicamente sotto forma di calore e lavoro. Allora il primo principio può essere formulato come segue: "Esiste una proprietà del sistema che si definisce Energia interna (U) ed è tale che, se il sistema è chiuso, la si può variare scambiando lavoro e/o calore con l'ambiente." La ΔU, essendo una funzione di stato, è indipendente dalle modalità con cui si opera la variazione e dipende unicamente dalla differenza delle Energie interne dello stato finale e iniziale. ΔU = Uf - Ui (1) Esempio: Consideriamo la combustione dell'ottano (componente principale della benzina) 2 C8H18(l) + 25 O2(g) à 16 CO2(g) + 18 H2O(l) Due quantità uguali di benzina: (1) usata come combustibile (bruciata) (2) usata come carburante (nel motore a scoppio) Se sono uguali gli stati iniziali (T, P e quantità di benzina) e se sono uguali gli stati finali (T, P e quantità di CO2 e H2O prodotte) nei due casi, si ha che ΔU(1) = ΔU(2). Per il primo principio ΔU = Q + W (questa equazione verrà descritta nella pagina successiva); ciò significa che Q1> Q2 perché nel secondo caso (motore a scoppio) si produce anche lavoro (W). Il Primo Principio (formulazione matematica) Nella formulazione del primo principio, abbiamo affermato che vi sono due meccanismi per variare l'energia interna in un sistema chiuso: lavoro e calore. Tutto ciò può essere espresso matematicamente come segue: ΔU = Q + W (2) Ovvero la variazione di energia interna di un sistema (ΔU) è uguale alla somma delle quantità di calore e lavoro scambiate tra il sistema e l'ambiente. In cui vale la seguente convenzione sui segni: E. Angelini – DISAT – Politecnico di Torino -4-
Corso di Chimica – A.A. 2014-2015 + w lavoro compiuto sul sistema – w lavoro compiuto dal sistema + q calore ceduto al sistema – q calore ceduto dal sistema In un sistema isolato, che non può scambiare né lavoro, né calore con l'ambiente, la ΔU è inevitabilmente ZERO. Pertanto, un'implicazione del primo principio è la seguente: "L'energia interna di un sistema isolato è costante", che è un modo diverso ma analogo della definizione precedente. Si tenga presente che l'Universo è il più grande dei sistemi isolati. L'energia interna si misura nei calorimetri, o meglio nelle bombe calorimetriche, utilizzando la seguente formula: ΔU = n • c • Δt = QV Dove n è il numero di moli della sostanza in esame, c è il calore specifico molare, Δt è la variazione di temperatura subita dalla sostanza. Un importante calorimetro è la "Bomba di Mahler" con cui si determina il calore di sostanze solide o liquide. Essa consiste in un piccolo recipiente di acciaio a pareti robuste, ermeticamente chiuso, in cui è posta una piccola capsula di porcellana contenente la sostanza in esame; nel recipiente viene immesso ossigeno puro e la combustione viene innescata mediante una resistenza E. Angelini – DISAT – Politecnico di Torino -5-
Corso di Chimica – A.A. 2014-2015
elettrica a contatto con la sostanza. Facendo passare corrente elettrica nella spiralina della
resistenza elettrica, questa si arroventa e brucia, provocando la rapida combustione del composto.
Il calore della reazione viene assorbito da una quantità nota di acqua, in cui è immersa la
"bomba".
Nota, mediante taratura, la capacità termica C del calorimetro e tenendo conto che il calore
specifico dell’acqua è uguale a 4,184 J·g-1·°C-1, possiamo risalire alla quantità di calore messa in
gioco nella reazione dall'espressione:
in cui:
m = massa in grammi dell'acqua presente all'interno del calorimetro
C = capacità termica del calorimetro
T1 = temperatura iniziale dell'acqua
T2 = temperatura finale dell'acqua
La capacità termica del calorimetro tiene conto del fatto che anche le sue parti componenti
(contenitore, termometro, agitatore, ecc.) assorbono calore. E' necessario pertanto predeterminare
la capacità termica del calorimetro, cioè determinare la quantità di calore necessaria per innalzare
di 1°C la temperatura di tale sistema: essa si può ottenere facendo svolgere nell'apparecchio una
reazione la cui tonalità termica sia nota, oppure facendo passare una quantità nota di corrente
elettrica, che viene dissipata come calore all'interno dello strumento.
Si definisce CALORE SPECIFICO di una sostanza è la quantità di energia che occorre usare per
far aumentare di un grado Celsius la temperature di un grammo di sostanza.
Il calore specifico si potrebbe però anche esprimere in quest'altro modo:
La quantità di energia che un grammo di sostanza cede all’ambiente raffreddandosi di un grado.
Alcune sostanze, come ad esempio i metalli, si scaldano molto velocemente e altrettanto
velocemente cedono all'ambiente il calore precedentemente accumulato. Essi hanno un basso
calore specifico, cioè occorre poca energia per scaldarli.
Altre sostanze, come ad esempio l'acqua, hanno un calore specifico molto alto. Ciò significa che
devo usare molta energia per scaldare l'acqua, ma anche che una volta che l'acqua si è scaldata,
tende a mentenere calore per molto tempo.
Non è un caso se il radiatore dell'automobile è fatto da sottili lamine di metallo. Il liquido
"trasporta" il calore in eccesso del motore al radiatore che si scalda rapidamente, ma anche molto
rapidamente cede calore all'ambiente (l'aria che arriva dall'esterno). In questo modo si riesce a
disperdere l'eccesso di calore del motore in modo molto più efficiente.
Calore specifico isobaro di alcune sostanze
Sostanza Stato
J/(kg·K)
Alluminio solido 880
Acciaio inox solido 502
Acqua liquido 4186
Acqua (Ghiaccio) solido (0 °C) 2090
E. Angelini – DISAT – Politecnico di Torino -6-Corso di Chimica – A.A. 2014-2015 Anidride carbonica liquido 838 Aria (secca) gassoso 1005 Aria (100% umidità relativa) gassoso ~ 1030 Azoto gassoso 1042 Berillio solido 1824 Diamante solido 502 Elio gassoso 5190 Etanolo liquido 2460 Ferro solido 444 Glicerina liquido 2260 Grafite solido 720 Idrogeno gassoso 14435 Litio solido 3582 Mercurio liquido 139 Olio liquido ~ 2000 Ossigeno gassoso 920 Oro solido 129 Ottone solido 377 Piombo solido 130 Polistirene solido 1450 Rame solido 385 Silice (fuso) solido 703 Silice gassoso 2020 Stagno solido 228 Zinco solido 388 Condizioni standard Per i solidi il valore coincide col calore specifico a volume costante IL PRIMO PRINCIPIO (EQUIVALENTE MECCANICO DEL CALORE) Il sistema non fa distinzione fra le due forme di energia, lavoro e calore. Una stessa variazione dell'energia interna si può ottenere trasferendo o solo lavoro o solo calore. Si può dare infatti una definizione meccanica della quantità di calore in termini di lavoro necessario per produrre la stessa variazione dell'energia interna, apprezzabile ad esempio come aumento di temperature prodotto in un sistema adiabatico. In termodinamica un sistema adiabatico è un sistema chiuso che non può scambiare calore con l'ambiente esterno, può invece scambiare lavoro. 1 caloria = 4.184 Joule È questo il cosiddetto equivalente meccanico della caloria, dimostrato da Joule. E. Angelini – DISAT – Politecnico di Torino -7-
Corso di Chimica – A.A. 2014-2015 UNA FUNZIONE DI STATO "DI COMODO": L'ENTALPIA (H) Essendo la variazione di energia interna uguale al calore scambiato a volume costante la formula: ΔU = n • c • Δt = QV non è applicabile alle numerosissime reazioni chimiche che avvengono a pressione costante, e, nelle quali l'energia non si trasforma completamente in calore, ma anche in lavoro. Consideriamo, allora, una reazione chimica (sistema chiuso, P = costante) in cui il lavoro compiuto dal sistema sia solo lavoro di volume. ______________________________________________________________________________________________________________________________ Lavoro di volume Ad es., consideriamo la reazione dello zinco con acido cloridrico a pressione costante. La pressione costante dell'atmosfera è stata sostituita da un pistone e da due pesi che esercitano una pressione equivalente. a) All'inizio della reazione, la pressione atmosferica è bilanciata dal pistone e dai pesi, cosicché all'interno del recipiente la pressione è costante. b) Nella reazione viene prodotto idrogeno; questo porta a un aumento di volume; i pesi e il pistone sono spinti verso l'alto. Il lavoro viene compiuto dal sistema verso il pistone e contro la forza-peso. Zn + 2 HCl à ZnCl2 + H2 Il lavoro di volume (WV) è una forma di energia legata alla variazione del volume occupato dal sistema di reazione. Se la variazione di volume avviene a pressione costante WV = PΔV • Se durante la reazione ΔV > 0, si ha w < 0; il sistema deve spendere parte della sua energia per compiere l'espansione che è contrastata dalla pressione, ovvero, come già indicato, il lavoro è negativo se compiuto dal sistema (ed è quello che avviene nell'esempio). • Se viceversa durante la reazione ΔV < 0, si ha w > 0; il sistema guadagna energia (fornita dall'ambiente), ovvero, come già indicato, il lavoro è positivo se compiuto sul sistema. • Se invece nella reazione ΔV = 0, si ha w ~ 0, per i solidi e i liquidi puri, mentre per i gas ideali w = RTΔn(g). • Nel calcolo del lavoro svolto da un gas che si espande, in genere si ottiene un risultato la cui unità di misura è espressa come prodotto tra le unità di misura di volume e pressione, cioè litri•atm; dal momento che è bene esprimere il lavoro utilizzando l'unità di misura del SI, cioè in E. Angelini – DISAT – Politecnico di Torino -8-
Corso di Chimica – A.A. 2014-2015 joule, è necessario effettuare le opportune conversioni: 1 litro•atm = 1 dm3 • 101325 Pa = 0,001 m3 • 101325 Pa = 102,3 J. Quindi, se nella reazione ΔV = 0, si ha W ~ 0, allora la relazione (2) diventa ΔU = QV Se, invece, la trasformazione avviene a P = costante (come generalmente accade in laboratorio – Processo isobaro), questo lavoro (w) è PΔV (lavoro di espansione); allora la (2) diventa: ΔU = Q - PΔV (8.3) con l'avvertenza che: Q è il calore che il sistema assorbe dall'ambiente, PΔV (w) è il lavoro compiuto dal sistema sull'ambiente; Q è positivo (+Q) perché significa guadagno di energia; PΔV (w) è preso con il segno (−PΔV) perché, essendo il lavoro fatto dal sistema, significa perdita di energia. Da cui: Q = ΔU + PΔV Definiamo ora una nuova funzione di stato, l'Entalpia (H), tale che la quantità U + PV viene considerate un’unica grandezza, cioè: H = U + PV (4) Dalla (8.4) si ha che, per una trasformazione in un sistema chiuso, a P = costante: ΔH = ΔU + PΔV (5) E, sostituendo q - PΔV per ΔU, otteniamo infine: ΔH = Qp (6) Tale grandezza esprime la differenza di entalpia che, a differenza del calore ha il vantaggio di essere una funzione di stato: essa, cioè, come l’energia interna U, dipende solo dallo stato iniziale e finale del sistema. Pertanto possiamo dire che: La variazione di entalpia ΔH indica il calore sviluppato o assorbito in una reazione, a condizione che questa venga condotta a pressione costante E, siccome essa è una funzione di stato, allora può essere espressa dalla relazione: ΔH = Hprodotti − Hreagenti (7) Inoltre, essendo, come tutte le funzioni termodinamiche, una grandezza estensiva, occorre sempre specificare la massa di materia interessata alla trasformazione. Nel caso di reazioni chimiche ci si riferisce sempre a una mole del composto che interessa e pertanto essa si misura in Kcal/mol o in KJ/mol , secondo il Sistema Internazionale. Infine, essendo, a pressione costante, la quantità di calore fornita per andare da T1 a T2 pari a: Qp = n•Cp•(T2 – T1) E. Angelini – DISAT – Politecnico di Torino -9-
Corso di Chimica – A.A. 2014-2015 dove n sono le moli e cp il calore specifico a pressione costante. Allora dalla definizione di entalpia, segue che: ΔH = Qp = n•Cp •(T2 – T1 ) Quindi in una trasformazione a pressione costante, detta ISOBARA, il calore scambiato è uguale alla variazione di entalpia e si può misurare con questa formula. Da queste considerazioni, tutte ricavate per via sperimentale, si deduce che l’energia interna e l’entalpia sono grandezze simmetriche che si utilizzano, rispettivamente, in caso di volume o pressione costante. ENTALPIA DI FORMAZIONE Come già sottolineato nel precedente paragrafo l’entalpia è una funzione di stato. In natura moltissimi processi avvengono a P = cost. A seconda del processo, il calore scambiato prende il nome dal tipo di processo, ad es.: entalpia di transizione, di combustione,… Ora, se in una reazione chimica consideriamo come stato iniziale l'insieme dei reagenti e come stato finale l'insieme dei prodotti, dovremmo poter calcolare una entalpia di reazione ΔHr, eseguendo una sottrazione fra la somma delle entalpie dei prodotti e la somma delle entalpie dei reagenti. Purtroppo però non siamo in grado di conoscere i valori assoluti di entalpia , cioè i valori di H da attribuire a ciascuna specie chimica che partecipa alla reazione, in quanto possiamo solo determinare valori di ΔH. In altre parole, siamo in grado di misurare solo la variazione di entalpia legata a certe trasformazioni . Insomma, quello che normalmente viene proposto nei testi come valore dell'entalpia dei vari composti chimici è, in realtà, una ΔH; essa è, infatti, la cosiddetta entalpia standard di formazione ΔH0f dei composti chimici. ΔH0f è la variazione di entalpia associata alla trasformazione degli elementi quando questi passano dai loro stati standard, al composto considerato, isolato nel suo stato standard. Esempio: formazione del metano a partire dal carbonio (grafite) e dall’idrogeno C (sol.) + 2H2 (gas) à CH4 (gas) ΔH0f = -74.8 kJ/mole a 25°C Il segno negativo indica che nella combustione viene rilasciata energia. Questa energia viene fornita come calore per ogni mole di CH4 che viene prodotta. Poiché l'entalpia è una funzione di stato che dipende da P, T e n, la variazione di entalpia in una reazione chimica (ΔH) dipende a sua volta da • natura della sostanza • temperatura e pressione (poco) alle quali avviene la reazione • quantità (massa o numero di moli) dei reagenti e dei prodotti coinvolti • stato di aggregazione in cui si trovano reagenti e prodotti Inoltre, poiché l'entalpia è una funzione di stato, invertendo il senso di una reazione, l'entalpia di reazione cambia semplicemente di segno. CH4 (gas) à C (sol.) + 2H2 (gas) ΔH0f = +74.8 kJ/mole a 25°C STATO STANDARD E. Angelini – DISAT – Politecnico di Torino - 10 -
Corso di Chimica – A.A. 2014-2015 Lo stato standard è definito da queste due condizioni: • Ci si riferisce ad una mole di sostanza (entalpie molari, espresse in kJ/mol) • Per ogni temperatura si definisce lo stato standard di una sostanza, caratterizzato dalla pressione di 1 atm (101325 Pa) e dallo stato di aggregazione in cui si trova normalmente la sostanza alla temperatura data e alla P di 1 atm. Quindi, per convenzione: • le entalpie di formazione tabulate si riferiscono alla formazione di una mole di sostanza pura nel suo stato standard a 25°C (298,15 K) partendo dagli elementi costitutivi nei loro stati standard. • l'entalpia standard di formazione a 298 K (ΔH0f 298) di tutte le specie elementari è uguale a zero, perché di fatto non avviene alcuna trasformazione! E. Angelini – DISAT – Politecnico di Torino - 11 -
Corso di Chimica – A.A. 2014-2015 ENTALPIA DI REAZIONE Conoscendo dunque il valore di ΔH0f per ogni singola specie chimica e sapendo che le funzioni di stato possono essere addizionate, è possibile determinare l'entalpia standard di reazione ΔH0r associata a una data reazione chimica: la ΔH0r si ottiene dalla somma algebrica delle entalpie di formazione di ogni molecola partecipante alla reazione, considerando che i prodotti addizionano un termine ΔH0f e i reagenti sottraggono un termine ΔH0f. In formula : ΔH0r = ΔH0f prodotti − ΔH0f reagenti. L'entalpia standard di formazione dei composti chimici è sempre riferita alla formazione di una mole di composto, perciò è sempre espressa utilizzando come unità di misura kJ/mol. Questo implica che • nel calcolo dell'entalpia di reazione, i valori dell'energia standard di formazione di ogni composto devono essere sempre moltiplicati per il coefficiente stechiometrico corrispondente. FORME DI ENTALPIA Esistono vari tipi di entalpia: Entalpia di transizione Entalpia di atomizzazione entalpia di soluzione Entalpia di combustione Entalpia di neutralizzazione entalpia di legame Entalpia di formazione Entalpia di atomizzazione entalpia di reticolo Entalpia di reazione Entalpia di transizione ............. Esempi Entalpia di transizione (nelle trasformazioni fisiche) H2O (liq.) à H2O (gas) ΔHvap = + 40.7 kJ/mole (Vaporizzazione) H2O (sol.) à H2O (liq.) ΔHfus = + 6 kJ/mole (Liquefazione) Entalpia di combustione (particolarmente utili per fare confronti tra i diversi combustibili) CH4 (gas) + 2O2 (gas) à CO2 (gas) + 2H2O (liq.) ΔHcomb. = - 890.4 kJ/mole a 1 atm e 25 °C. Il segno negativo indica che nella combustione viene viene rilasciata energia . C6H12O6 (sol.) + 6O2 (gas) à 6CO2 (gas) + 6H2O (liq.) ΔHcomb = - 2816 kJ/mol questa energia corrisponde a 15.6 kJ per g di glucosio Questa reazione è la sorgente di energia degli animali i quali usano la respirazione per sfruttare le risorse energetiche fornite dalla digestione e convertirle in attività metaboliche (attività cardiocircolatoria, respiratoria, di termoregolazione,...). LA LEGGE DI HESS Le entalpie di reazione in condizioni standard vengono indicate con ΔH°r . Quando non è possibile determinare sperimentalmente il ΔH°f di un composto o il ΔH°r di una reazione, si può applicare la legge di Hess: il ΔH°r di una reazione chimica è dato dalla differenza tra la sommatoria dei ΔH°f dei prodotti e la sommatoria dei ΔH°f dei reagenti, essendo ciascun ΔH°f moltiplicato per il coefficiente stechiometrico di reazione , cioè: E. Angelini – DISAT – Politecnico di Torino - 12 -
Corso di Chimica – A.A. 2014-2015 Le applicazioni della legge di Hess, numerose e di grande importanza pratica, si basano sulla proprietà di funzione di stato dell'entalpia dei sistemi, per cui il ΔH ° di una trasformazione non dipende dal cammino percorso. Si può sempre immaginare, infatti, di passare da uno stato 1 a uno stato 2 di un sistema chimico attraverso reazioni intermedie che consentono il calcolo del ΔH°f di alcuni composti o del ΔH ° di reazione, quando questi non possono essere determinati per via diretta. Per esempio, si calcoli il ΔH ° della reazione: sapendo che per la reazione: e che per la reazione: Poiché la reazione desiderata si ottiene sommando la (2) alla (1), il ΔH ° della reazione in esame si calcolacon la stessa operazione, cioè: REAZIONI ESOTERMICHE E ENDOTERMICHE L'aver introdotto la funzione di stato Entalpia, porta alla conclusione che: "In un sistema chiuso, la variazione di Entalpia è pari alla quantità di calore scambiato, poste due restrizioni: l'unica forma di lavoro possibile è il lavoro di espansione e P = costante." Per scambi di calore a T = costante, in base alla direzione del flusso di calore (il segno di ΔH), le reazioni chimiche si suddividono in: Esotermiche il sistema libera calore q < 0 ΔH < 0 Isotermiche nessuno scambio di calore q = 0 ΔH = 0 Endotermiche il sistema assorbe calore q > 0 ΔH > 0 In altre parole, durante ogni trasformazione chimica alcuni legami si rompono e se ne formano di nuovi: Per rompere un legame chimico è necessario fornire energia, mentre quando si forma un legame si libera energia. Soltanto quando i legami che si formano sono più forti di quelli che di quelli che si rompono si libera energia, cioè la reazione è esotermica, sarà, invece, endotermica nel caso contrario. E. Angelini – DISAT – Politecnico di Torino - 13 -
Corso di Chimica – A.A. 2014-2015
Reazioni
esotermiche
Reazioni
endotermiche
Si
formano
molecole
più
stabili
con
legami
più
forti
Si
formano
molecole
meno
stabili
con
legamipiù
deboli
L'energia
potenziale
del
sistema
diminuisce
e
si
produce
calore,
che
fluisce
verso
l'ambiente
L'energia
potenziale
del
sistema
aumenta
a
spese
del
calore
assorbito
dall'ambiente
REVERSIBILITÀ E IRREVERSIBILITÀ TERMODINAMICHE
Si ha un processo termodinamicamente reversibile, quando una trasformazione viene condotta in
modo che il sistema passi attraverso stadi intermedi che differiscano da stati di equilibrio per
quantità infinitesime.
In pratica, per operare una trasformazione reversibile, si devono alterare le condizioni in modo
sufficientemente lento da consentire al sistema di adattarsi alle nuove condizioni, ad es.:
Supponiamo di avere due recipienti contenenti acqua, il primo recipiente è aperto, mentre il
secondo è chiuso con un tappo.
Supponiamo di scaldare entrambi i recipienti favorendo l'evaporazione dell'acqua.
Nel caso del recipiente aperto il processo di evaporazione è irreversibile, perché le molecole
d'acqua fuoriescono dalla fase liquida passando in fase di vapore e si disperdono nell'ambiente.
Nel caso del recipiente chiuso, invece, durante l'evaporazione le molecole d'acqua rimangono
intrappolate tra la superficie del liquido e il tappo, perciò, raffreddando il sistema, è possibile far
passare le molecole dalla fase di vapore alla fase liquida ripristinando la situazione iniziale: in
questo caso il processo risulta reversibile.
Se trasformiamo un sistema reversibilmente da uno stato iniziale A ad uno stato finale B,
possiamo poi riportare il sistema dallo stato B a quello A attraverso la trasformazione inversa.
Tuttavia, l'esperienza ci insegna che i processi naturali evolvono spontaneamente (nel tempo) in
una determinata direzione, ad es.:
E. Angelini – DISAT – Politecnico di Torino - 14 -Corso di Chimica – A.A. 2014-2015 L'acqua di una cascata cade spontaneamente da una quota maggiore a una quota minore. Un gas si espande spontaneamente fino ad occupare tutto il volume a disposizione. Un corpo caldo si raffredda spontaneamente fino a raggiungere l'equilibrio termico con l'ambiente circostante. Il comune sale da cucina si scioglie spontaneamente in acqua. Il carbonio si combina spontaneamente con l'ossigeno dell'aria per dare il diossido di carbonio. La trasformazione in ruggine di un oggetto di ferro esposto all'aria e all'umidità avviene spontaneamente. Le trasformazioni di questo tipo non possono mai essere invertite completamente e per questo motivo, I processi naturali sono definiti termodinamicamente irreversibili. L'acqua precipitata da una cascata non può spontaneamente risalire alla quota iniziale; per farlo si deve impiegare dell'energia (ad es., ricorrendo a una pompa). Un gas che diffonde da un recipiente a un altro attraverso una valvola di comunicazione, non fa spontaneamente il processo inverso. Il calore non passa spontaneamente da un corpo freddo a uno caldo: perché ciò accada occorre utilizzare una macchina che "crei" il freddo, ad es. un frigorifero. Il comune sale da cucina non si separa spontaneamente dall'acqua se non si fornisce dall'esterno dell'energia, per es., per far evaporare l'acqua. Per ottenere nuovamente il carbonio e l'ossigeno dal diossido di carbonio occorre fornire energia per rompere i legami che tengono insieme i suoi atomi. Per riottenere il metallo contenuto nella ruggine occorre un processo chimico analogo a quello con cui si ottiene un metallo dai suoi minerali, e ciò richiede energia. Dietro al concetto di irreversibilità vi è di fatto "l'impossibilità di riportare un sistema, che ha subìto una trasformazione spontanea, dallo stato finale a quello iniziale, senza provocare mutamenti in qualche parte dell'universo." Questo punto è di fondamentale importanza e fa la differenza fra trasformazione reversibile e irreversibile. Un processo spontaneo può essere invertito intervenendo dall'esterno, ad esempio spendendo lavoro e/o energia; tuttavia, in qualche modo si produce inevitabilmente una variazione in qualche altro sistema. IL SECONDO PRINCIPIO: LA DOMANDA "Cos'è che determina la direzione di un processo spontaneo?" "In che modo si può prevedere se un processo è spontaneo?" Il motore che spinge i sistemi verso uno stato stabile, di equilibrio, è la tendenza verso uno stato di minima energia potenziale. Una valanga, ad es., raggiunge la situazione di massima stabilità quando si arresta nel fondovalle. Nelle trasformazioni chimiche spontanee, in generale, le cose vanno allo stesso modo. Prendono parte a una reazione chimica le sostanze che si trovano in una condizione instabile e che in tal modo tendono spontaneamente a raggiungere una condizione finale stabile. Ora, sappiamo dal 1° principio che l'energia dell'universo è costante. Quindi, se il criterio della minima energia fosse valido, dovremmo comunque aspettarci di veder aumentare l'energia da qualche parte, diversa dal sistema. E in tal caso, non sarebbe altrettanto corretto prendere in considerazione l'aumento di energia dell'ambiente, come criterio di spontaneità? In tal caso soltanto le reazioni esotermiche dovrebbero avvenire spontaneamente. Sebbene la maggior parte delle reazioni spontanee sia in effetti esotermica, si conoscono tuttavia numerose reazioni spontanee che avvengono a temperatura ambiente con assorbimento di calore, che sono cioè reazioni endotermiche, per es.: E. Angelini – DISAT – Politecnico di Torino - 15 -
Corso di Chimica – A.A. 2014-2015 MgCl2(aq) + Na2CO3(aq) à MgCO3(s) + 2 NaCl(aq) ΔH° = +25kJ È probabile che dovremo cercare la risposta altrove. IL SECONDO PRINCIPIO: LA RISPOSTA La risposta va infatti cercata in una "nuova" funzione di stato, l'Entropia (S). Partiamo da una premessa di natura storica. Dal 1° principio si deduce anche che non può esistere una macchina capace di creare energia, e proprio di macchine termiche e del loro rendimento si occupava, nel 1824 Sadi Carnot quando si imbattè senza saperlo in quello che poi, rielaborato da Kelvin e da Clausius, diventerà nel 1851 il 2° principio della termodinamica. Esso afferma, nella prima formulazione di Kelvin, che è impossibile realizzare un trasferimento spontaneo di calore da un corpo più freddo a uno più caldo e, nella seconda formulazione di Clausius, che è impossibile che tutto il calore di una sorgente si trasformi in lavoro. In sintesi, tutte le volte che si produce lavoro dal calore (in tutte le macchine termiche), contemporaneamente ha luogo un passaggio di calore dal corpo più caldo a quello più freddo, mai in senso opposto. Quindi la produzione di lavoro, ce lo conferma la nostra esperienza quotidiana, è accompagnata in tutte le trasformazioni energetiche da un riscaldamento, c'è allora una tendenza dell'universo verso la "forma calore" e il calore è una forma degradata di energia perché non si lascia riconvertire totalmente in lavoro. In conclusione, mentre il 1° principio si occupa del bilancio generale dell'energia, il 2° si occupa delle trasformazioni di energia, del suo uso e della sua tendenza naturale ad andare verso forme degradate, non utilizzabili: ciò che diminuisce nell'universo allora non è l'energia, ma la sua capacità di compiere lavoro. Torniamo ora all'entropia. Una delle possibili formulazioni del Secondo Principio è la seguente: "In una trasformazione spontanea l'Entropia totale del sistema e dell'ambiente aumenta." ; o, anche, il che è la stessa cosa: "In ogni processo naturale l'Entropia dell'Universo aumenta." L' Entropia è spesso indicata come misura del disordine di un sistema: Maggior disordine, maggior entropia e viceversa. Dobbiamo tuttavia cercare di dare una definizione un po' "meno entropica" della nostra nuova funzione di stato. ENTROPIA: IL CONCETTO STATISTICO All'entropia (S ) possiamo attribuire un significato statistico di "probabilità". Per cercare di semplificare, abbiamo bisogno di un paio di termini accessori - microstato e macrostato – e magari di un esempio pratico. Iniziamo col definire S come "il numero di microstati di un determinato macrostato". Per microstato possiamo intendere una qualsiasi configurazione equiprobabile degli elementi di un sistema. Per macrostato intendiamo invece una categoria di configurazioni (un insieme di microstati) che rispondano a determinati requisiti. Per semplicità, supponiamo di avere un sistema costituito da un recipiente contenente tre particelle, riconoscibili per il diverso colore: rosso, verde e blu. Se suddividiamo idealmente il recipiente in due parti dello stesso volume, le tre particelle potranno distribuirsi in 23 = 8 configurazioni diverse: otto microstati, tutti equiprobabili. Questi microstati possono essere suddivisi in due categorie: • Macrostato 1: Tutte le particelle sono da una stessa parte del recipiente (microstati 4 e 8). E. Angelini – DISAT – Politecnico di Torino - 16 -
Corso di Chimica – A.A. 2014-2015
• Macrostato 2: Le particelle sono distribuite 2:1 o 1:2 tra la parte sinistra e la parte destra
(microstati 1,2,5 e 3,6,7).
Poiché il Macrostato 2 si può realizzare attraverso sei microstati diversi, tutti equiprobabili
(contro i due del Macrostato 1), esso rappresenterà la configurazione più probabile del sistema.
In base alla definizione di S, diremo che il Macrostato 2 ha un'entropia più elevata del Macrostato
1.
L'esempio delle tre particelle non rende piena giustizia al "potere" dell'entropia. Già supponendo
di avere 10 particelle, i microstati possibili salirebbero a 210 = 1024; resterebbero 2 i microstati
del Macrostato "tutte le particella da un lato", mentre 20 dei 1024 rappresenterebbero il
Macrostato "distribuzione 9:1 e 1:9". Il Macrostato "5 particelle su ciascun lato" si realizzerebbe
invece attraverso 252 microstati; quindi una distribuzione esattamente uniforme avrebbe una
probabilità 25 volte maggiore di una distribuzione 9:1 e la probabilità di trovare tutte le particelle
da un lato sarebbe addirittura 126 volte minore.
Immaginate ora quale possa essere la situazione delle molecole dell'aria contenuta in una comune
stanza: circa 1027 molecole!
Qualche esempio.
• Una zolletta di zucchero è un sistema certamente molto ordinato. Se però tale zolletta viene
posta in un bicchiere d'acqua, essa passa spontaneamente in soluzione, che è un sistema con un
più alto grado di disordine.
• Nei passaggi di una sostanza dallo stato solido a quello liquido ed infine, a quello aeriforme
aumenta il grado di disordine.
• Si ha un aumento di disordine nelle reazioni di decomposizione, quando si passa da molecole
formate da tanti atomi, cioè grandi, a molecole formate da pochi atomi, cioè piccole.
• Si ha un aumento di disordine nelle reazioni in cui almeno uno dei prodotti è gassoso
Quesiti:
• Perché le molecole di un gas tendono ad occupare spontaneamente tutto lo spazio a loro
disposizione?
• Giocando ai dadi, qual è il macrostato più probabile? Perché?
ENTROPIA: IL CONCETTO TERMODINAMICO
• L'entropia è una funzione di stato e pertanto la sua variazione dipende solo dagli stati finale (B)
e iniziale (A) della trasformazione:
(8)
• A un qualsiasi processo è sempre associata una variazione di entropia, che indichiamo con
ΔStotale. Essa è, in generale, la somma di due contributi, quello esterno, che indichiamo con
ΔSambiente e quello interno, che indichiamo con ΔSambiente:
E. Angelini – DISAT – Politecnico di Torino - 17 -Corso di Chimica – A.A. 2014-2015
ΔStotale = ΔSsistema + ΔSambiente (9)
• Il ΔSambiente è legato al calore scambiato tra l'ambiente e il sistema durante il processo, e si ricava
dal rapporto tra il calore scambiato nel processo e la temperatura a cui avviene lo scambio:
(10)
• Il ΔSsistema non implica scambio di calore ma corrisponde, invece, a una vera e propria creazione
di entropia, cioè di disordine, perché è collegato al modo in cui il sistema si dispone, ovvero alla
sua configurazione.
Poiché l'entropia dell'universo tende spontaneamente ad aumentare e che a qualunque processo
spontaneo (ovvero trasformazione irreversibile) corrisponde un aumento di disordine, allora
ΔStotale = ΔSsistema + ΔSambiente > 0 (11)
Al contrario, in una trasformazione ciclica reversibile, dato che gli stati iniziale e finale
coincidono, la variazione di entropia è nulla.
È quindi evidente che i due tipi di trasformazione hanno effetti diversi sullo stato dell'ambiente.
A questo proposito è di fondamentale importanza tener presente che nel corso di un processo
irreversibile, la qualità dell'energia si degrada e perde parte della propria capacità di produrre
lavoro. In definitiva, l'entropia può essere vista come un indice dell'esaurirsi della idoneità di un
sistema a produrre trasformazioni.
Una ulteriore formulazione del secondo principio è difatti la seguente:
"Nei processi reversibili la variazione complessiva di entropia del sistema e dell'ambiente è nulla;
nei processi irreversibili è positiva."
Ovvero, "In un processo spontaneo si ha sempre un aumento dell'entropia totale".
• Per un processo reversibile: ΔStot = ΔSsist + ΔSamb = 0
• Per un processo irreversibile: ΔStot = ΔSsist + ΔSamb > 0
Insomma quando si fanno considerazioni che coinvolgono l'entropia è necessario tener conto non
solo del sistema, ma anche di quello che accade nell'ambiente .
CHE COS'È REALMENTE SPONTANEO?
In base all'esperienza quotidiana, ciascuno di noi è in grado di fornire esempi corretti di processi
naturali spontanei e di fare previsioni più o meno sensate sulla loro naturale evoluzione
temporale.
Ad esempio, se il caffè che abbiamo appena versato nella tazzina è bollente, sappiamo che basterà
attendere qualche istante perché si raffreddi e diventi bevibile; dopo un acquazzone, ci aspettiamo
che la strada prima o poi si asciughi; così come dopo una nevicata, sappiamo che la neve presto o
tardi si scioglierà.
Siamo quindi capaci di riconoscere la natura spontanea di processi come il trasferimento di calore
da un corpo più caldo ad uno più freddo, la fusione del ghiaccio e l'evaporazione dell'acqua.
Giunti
a
questo
punto,
siamo
forse
anche
in
grado
di
dare
una
spiegazione
in
termini
entropici
della
spontaneità
di
questi
processi:
il
ghiaccio
fonde
spontaneamente
perché
le
molecole
dell'acqua
allo
stato
liquido
hanno
un'entropia
maggiore,
avendo
maggiore
libertà
di
movimento
e
allo
stesso
modo
possiamo
giustificare
l'evaporazione
spontanea
dell'acqua.
Ovvero
nei
passaggi
di
stato
si
ha
sempre
una
variazione
di
entropia
secondo
questo
ordine:
Ssolido
<
ΔSliquido
<
ΔSaeriforme
Ovvero,
quando
una
sostanza
si
trova
nello
stato
solido,
le
sue
particelle
sono
disposte
in
modo
ordinato,
con
scarsissima
possibilità
di
movimento,
quindi
l'entropia
di
un
solido
è
molto
bassa.
Quando
un
solido
passa
nella
fase
liquida,
le
sue
particelle
sono
molto
vicine,
ma
hanno
una
E. Angelini – DISAT – Politecnico di Torino - 18 -Corso di Chimica – A.A. 2014-2015 certa libertà di movimento e possono presentare ad ogni istante diverse configurazioni, quindi l'entropia di un liquido è sempre maggiore di quella di un solido. Infine, quando un liquido passa nella fase aeriforme, le sue particelle, a parità di numero di moli, occupano un volume considerevolmente più ampio e quindi hanno una libertà di movimento assai maggiore: l'entropia di un gas è assai maggiore di quella di un liquido. Ma se in una rigida giornata d'inverno osserviamo che l'acqua delle pozzanghere è gelata, cosa dobbiamo pensare? Che si è invertita la direzione di un processo spontaneo? Che in determinate condizioni il secondo principio perde di validità? Dov'è che stiamo sbagliando? È che stiamo considerando il processo limitatamente al sistema: l'acqua ghiacciata, nel caso specifico. Invece, dobbiamo anche valutare cosa è accaduto contemporaneamente nell'ambiente. Se la temperatura è sotto lo zero, l'acqua gela spontaneamente, ma il secondo principio conserva la propria validità. È vero che l'entropia del "sistema acqua" diminuisce, ma nel processo di congelamento vi è un trasferimento di calore dall'acqua all'ambiente. A causa del calore assorbito, l'entropia dell'ambiente aumenta più di quanto diminuisca quella dell'acqua, congelando: in questo modo il bilancio netto entropico risulta positivo. Anche gli esseri viventi apparentemente sfuggono al secondo principio della termodinamica ma ciò, oltre a essere impossibile, non è vero. Essi riescono a mantenere l’ordine interno e quindi entropia negativa, rilasciando nell’ambiente fattori entropici che compensano ampiamente l’ordine interno ed in modo che l’entropia ambiente-‐sistema cellula sia maggiore di 0. In sostanza -‐ e lo abbiamo appena definito quantitativamente nella pagina precedente -‐, il trasferimento di calore è uno dei meccanismi più comuni per aumentare l'entropia di un corpo, l'entropia del corpo al quale il calore viene ceduto. (Certo, alla luce di queste considerazioni, viene spontaneo domandarsi perché l'uomo si sia accanito così tanto con i motori termici, se il calore risulta una forma di energia così poco efficace ed efficiente). Comunque, per evitare il rischio di giungere a conclusioni quantomeno affrettate, formalizzeremo nell'ultimo paragrafo un criterio semplice e razionale per quantificare, sotto determinate assunzioni, il bilancio entropico totale di una trasformazione e quindi per valutarne la spontaneità: l'energia libera. LE VARIAZIONI DI ENTROPIA IN UN SISTEMA CHIMICO La variazione di entropia (ΔS) in una reazione chimica è data dalla differenza fra l'entropia dei prodotti e quella dei reagenti, ed è misurata in J/K, ma, se ci riferiamo ad una mole di sostanza, parliamo di entropia molare, misurata in J/mol•K. Pertanto l'entropia standard di reazione (ΔrS°) è data dalla somma algebrica delle entropie molari (S°) dei prodotti puri, riportate quindi con segno positivo, e dei reagenti puri, riportate con segno negativo, ognuna moltiplicata per il coefficiente stechiometrico presente nella reazione considerata: ΔrS° = S°prodotti − S°reagenti Nei calcoli relativi alla determinazione dell'entropia standard di reazione (ΔrS°) non si utilizzano i valori di ΔfS° delle singole sostanze, come siamo costretti a fare con l'entalpia, ma si fa ricorso semplicemente ai valori di S° delle singole sostanze. E' possibile stabilire il valore assoluto dell'entropia di una sostanza con metodi calorimetri; qui, tuttavia, è importante puntualizzare i E. Angelini – DISAT – Politecnico di Torino - 19 -
Corso di Chimica – A.A. 2014-2015 seguenti aspetti relativi a questo concetto: L'entropia molare standard, S°, d i una sostanza è l'entropia per mole della sostanza pura alla pressione di 1 atm (101,3 kPa) e a 298 K di temperatura. Le entropie sia degli elementi sia dei composti sono positive, cioè S° > 0 ; al contrario la variazione di entalpia standard di formazione degli elementi nelle loro forme stabili, ΔH°, è zero; quella dei composti può essere positiva o negativa. Pertanto, in un sistema chimico che dà luogo a una certa reazione chimica sono, in generale, lo stato di aggregazione dei reagenti e dei prodotti e la stechiometria della reazione stessa a determinare le variazioni di entropia che accompagnano la trasformazione chimica. In generale, si ha ΔS > 0 quando: • una soluzione viene diluita; • reagenti solidi o liquidi formano prodotti gassosi; • nel corso di una reazione, aumenta il numero delle moli delle specie gassose; • nel corso di una reazione aumenta il numero delle moli passando dai reagenti ai prodotti. Si ha, invece, ΔS < 0 nei processi inversi dei precedenti. Per esempio, la reazione esotermica: 3 H2(g) + N2(g) à2 NH3(g) ΔH° = −91,8 kJ comporta una diminuzione di entropia: ΔSreazione = ΔSprodotti − ΔSreagenti < 0 Infatti, tutte le specie chimiche sono gassose, ma la reazione avviene con diminuzione del numero di moli e quindi raggiunge uno stato di minor disordine finale. La reazione di decomposizione del carbonato di calcio (endotermica): CaCO3(s) à CaO(s) + CO2(g) ΔH° = +572 kJ comporta un aumento di entropia: ΔSreazione = ΔSprodotti − ΔSreagenti > 0 Infatti, il numero delle moli dei prodotti è maggiore di quello dei reagenti, ma, soprattutto, il fattore determinante è la comparsa di una fase gassosa ad alta energia. IL CRITERIO DI SPONTANEITÀ: LA FUNZIONE DI GIBBS (G) Come abbiamo appena visto, il criterio di spontaneità è dunque fissato dalla relazione: ΔStot = ΔSsist + ΔSamb > 0 (12) Il fatto è che, valutare la variazione di entropia che subiscono sistema ed ambiente nel corso di una trasformazione, è spesso poco pratico. Infatti, se è facile definire il sistema ed eseguire misure su quest'ultimo, la stessa cosa non vale per l'ambiente che, come abbiamo già visto, è definito come tutto ciò che circonda il sistema stesso. L'ideale sarebbe poter focalizzare l'attenzione su una qualche proprietà esclusiva del sistema, in grado di informarci sulla direzione spontanea del processo. Questa opportunità esiste ed è possibile definendo una nuova funzione di stato, detta energia libera di Gibbs (G), tale che: G = H - TS Analogamente all'entalpia e all'entropia, anche per l'energia libera G ciò che interessa è la variazione delle grandezze coinvolte, più che i loro valori assoluti, per cui possiamo scrivere: ΔG = ΔH - TΔS (13) Questa relazione, oltre a contenere grandezze facilmente misurabili perché riferite al sistema, fornisce un valido criterio di spontaneità delle reazioni chimiche a temperatura e pressioni costanti. Infatti è possibile dimostrare che, in tali condizioni sperimentali, una reazione chimica avverrà spontaneamente se si accompagna ad una variazione negativa della funzione di Gibbs: ΔG < 0 (14) E. Angelini – DISAT – Politecnico di Torino - 20 -
Puoi anche leggere

















































