Come studiare un genoma complesso - Costruirne la mappa
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Come studiare un genoma
complesso
1. Costruirne la mappa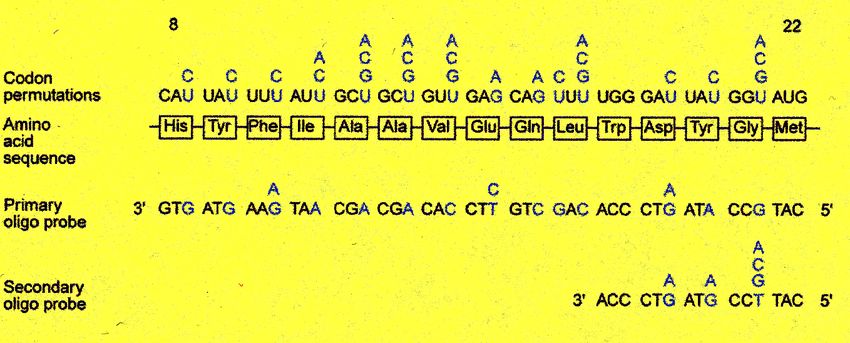
Costruzione della mappa di un cromosoma
•Obiettivo: ordinare in modo reciproco lungo il cromosoma geni
responsabili di fenotipi riconoscibili, o in generale qualsiasi sequenza
di DNA riconoscibile.
bello/brutto
alto/basso
Occhio azzurro/marrone
biondo/bruno
magro/grasso
ricco/squattrinato
Risultato

Strategie generali di mappaggio
• Mappaggio Genetico
• definizione: l’ordinamento di geni sui cromosomi in
accordo con la frequenza di ricombinazione
• il mappaggio genetico può essere utile prima che siano
disponibili sonde per il mappaggio fisico
• Mappaggio Fisico
• definizione: la determinazione della distanza fisica
tra i geni (espressa come coppie di basi del DNA)
usando tecniche citogenetiche e molecolari
• il mappaggio fisico è usato per comporre le sequenze
dei genomi complessi.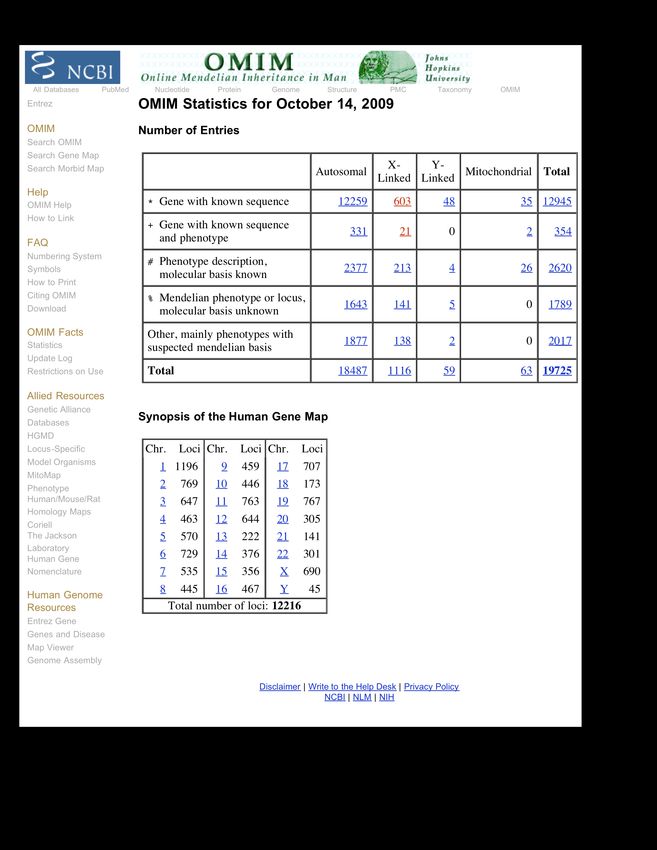
Per la costruzione di una qualsiasi mappa
sono necessari dei marcatori
(elementi facilmente riconoscibili)
•Le prime mappe allestite erano basate su criteri genetici; i primi marcatori
ad essere utilizzati sono stati geni ai quali fossero associati fenotipi
alternativi (alleli).
Le mappe basate sui geni non sono tuttavia molto dettagliate (non tutti
presentano forme alleliche diverse o facilmente riconoscibili, ed inoltre,
nei genomi complessi, i geni sono molto “diluiti” nel genoma.
•Marcatori di sequenza, non necessariamente genici, sono chiamati
marcatori di DNA. Come per i geni, i marcatori di DNA devono presentare
almeno due diverse forme alleliche, devono cioè essere polimorfici.
•Nel mappaggio fisico i marcatori possono essere le sequenze dei singoli
cloni.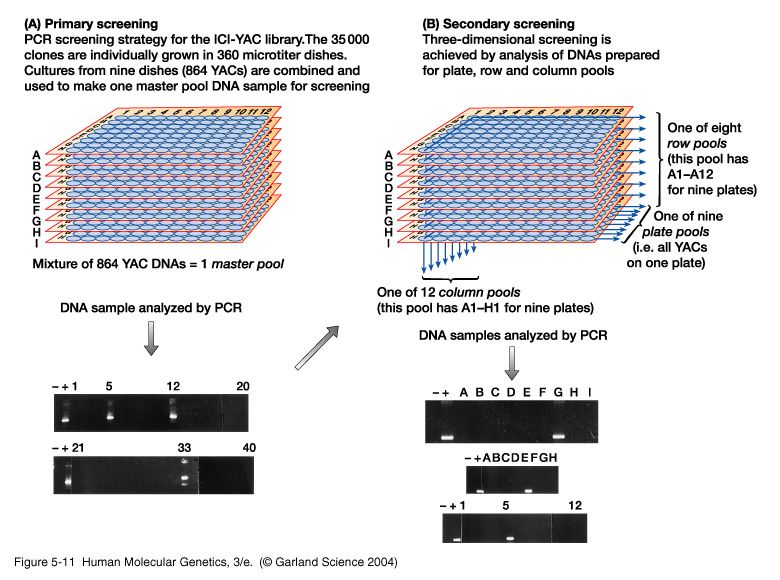
Tipi di mappe: mappe genetiche Mappe genetiche: si basano sulla frequenza di ricombinazione fra loci identificati attraverso marcatori di varia natura: fenotipo dell’individuo, fenotipo tissutale, fenotipo cellulare, fenotipo proteico, fenotipo del DNA. Sono la connessione fra una realta’ biologica e il genoma corrispondente, senza di loro spesso non si puo’ procedere,
Per il mappaggio genetico, qualora non si usino fenotipi
ma semplici sequenze di DNA, marcatori, queste devono
essere polimorfiche
RFLP-VNTR-SNP
I due alleli 1 e 2 si
differenziano per la
presenza/assenza di un sito
di restrizione nel cromosoma 1/2 1/2
GAGTTC 2/2 1/1
EcoRI EcoRI
500
500
1
350
GAATTC
EcoRI EcoRI 150
2 350 150

MARCATORI GENETICI
Tipo di N° loci Caratteristiche
Marcatore
RFLP >105 2 alleli marcatori,
potenzialmente eterozigosi massima 0,5.
Southern Blot o PCR. Facile
localizzazione fisica
Minisatelliti >104 Molti alleli altamente
potenzialmente informativi. Tipizzazione con
Sothern Blot. Facile
localizzazione fisica.
Tendono a localizzarsi vicino
ai telomeri
Microsatelliti >105 Molti alleli altamente
potenzialmente informativi. Si possono
tipizzare mediante PCR
(multiplex). Facile
localizzazione fisica.
Distribuiti lungo tutto il
genoma
SNP >105 Meno informativi. Possono
potenzialmente essere tipizzati su ampia
scala con appparacchiature
automaticheUna volta piazzati i “marcatori di DNA” sui cromosomi,
si può procedere alla costruzione della mappa genetica
mediante tecniche che si basano
sull’associazione genetica.
L’analisi di associazione si può effettuare in diversi modi:
•su specie come la drosofila o il topo mediante esperimenti
di incroci programmati;
•nell’uomo mediante lo studio dei pedigree.Analisi di linkage mediante RFLP
• Gli RFLP forniscono marcatori utili per tutti i cromosomi umani
• i geni malattia ( ) possono essere mappati cercando un
linkage tra un polimorfismo, ad esempio un RFLP e il fenotipo
malattia
Marcatore RFLP
A A’ probe Allele mutato
A’ A
allele normale
• il polimorfismo A/A’ è in linkage con il gene causa della malattia
AA AA’ A’A’
AA omozigote
AA’ eterozigote
A’A’ omozigoteUna volta piazzati i “marcatori di DNA” sui cromosomi,
si può procedere alla costruzione della mappa
genetica, inserendo geni di interesse, mediante
l’associazione genetica.
linkage del gene NF1
(neurofibromatosi) al
Locus 1 A a locus 2 ma non al locus 1;
NF1 B b (wild type)
Locus 2 C c
a A A a A
B B b b b
C C c c c
(sono mostrati solo i cromosomi paterni)Il pedigree mostra l’ereditarietà di una malattia genetica,
di cui non è noto né il gene né la posizione di mappa.
L’analisi dimostra che la malattia
ha una trasmissione autosomica
dominante.
Il gene malattia è in linkage con il marcatore, Ipotesi 0
l’obiettivo è determinare la fase tra il gene
malattia e il microsatellite M, determinando
M1
M2 X
Sano
Malato
la segregazione degli alleli di quest’ultimo
(M1-M2-M3....) nei membri della famiglia.
L’analisi del pedigree suggerisce
l’ipotesi 1 come la più probabile
e l’analisi sulla nonna materna
conferma definitivamente
l’ipotesi.Analisi di Linkage per la neurofibromatosi (NF1)
nf1
= NF1
Nf1
1
2
o
Nf1
2
nf1
1
nf1
2
nf1
2
Nf1
nf1
1
2
nf1
nf1
1
1
nf1
2
nf1
Southern blot 1
1 *
2
L’allele rappresentato dalla L’allele rappresentato dalla banda
banda 1 è diagnostico 2 è diagnostico
* = ricombinanteAncora terminologia Aplotipo o fase: l’ordine degli alleli di geni diversi sul cromosoma parentale. Se prendo in considerazione 3 loci (A, B e C) ognuno presente in eterozigosi (A1,A2; B1,B2; C1,C2) in un dato soggetto, a priori non posso sapere l’ordine sui due cromosomi parentali, che può essere: A1,B1,C1; A2,B2,C2; A1,B1,C2; A2,B2,C1; A1,B2,C1; A2,B1,C2; A1,B2,C2; A2,B1,C1; Nelle popolazioni naturali solo l’analisi della progenie mi dice quale era l’aplotipo dei parentali e solo nei grandi numeri. Le cose si complicano ulteriormente quando non disponendo di “fenotipi codominanti”, come i polimorfismi, posso seguire i loci solo con caratteri soggetti alla dominanza e recessivita’.
Linkage:fase
ATTENZIONE ALLA RICOSTRUZIONE DELLA FASE
Fase : combinazione degli alleli di una regione che deriva da ciascun
genitore.
Meiosi informative: sono quelle che danno informazioni sulla ricombinazione.
Il locus a (alleli 1, 2, 3 e 4) è in linkage con il locus malattia (M,m)
a1 M a1 M
a1 M a1 M
a1 m a1a1 a2a2 a2 m a1a2 a1a2 a1a2 a1a2 a1a2 a3a4
a2 m a2 m
a1 M
a1 m
a1a2 a1a2 a1a1 a1a4
a1 m
a2 M
non infor. non infor. infor. infor.Per la maggior parte dei genomi eucariotici la
mappa genetica deve essere controllata e
complementata da procedure di mappatura
alternative.
La mappatura fisica dei genomi.Il genoma umano
Creazione di librerie genomiche
Siti di restrizione potenziali
per un dato enzima
Digestione parzialeIl clonaggio
Quale vettore?
la ligazione
La trasformazioneSelezione dei cloni
Crescita
ricombinanti
clone ricombinanteScelta del vettore
V
VEET
TTTO
ORRE
E TAGLIA U
UTTIIL
LIIZ
ZZZO
O
DELL'INSERTO
PLASMIDI 0-10kb Clonaggio e amplIficazione di
sequenze di DNA
Espressione di sequenze geniche
→procarioti
→eucarioti
FAGI 20-25kb Costruzione di banche di DNA
Derivati da λ (LIBRERIE GENOMICHE)
Costruzione di banche di mRNA
(LIBRERIE DI cDNA)
COSMIDI 30-44kb Costruzione di LIBRERIE GENOMICHE
PAC (P1 derived artificial chromosome) 130-150kb Costruzione di LIBRERIE GENOMICHE
BAC (Bacterial artificial chromosome) Fino a 300kb Costruzione di LIBRERIE GENOMICHE
YAC (Yeast artificial chromosome) 0.2-2Mb Costruzione di LIBRERIE GENOMICHE
(chimerismo)Yeast Artificial Chromosome (YAC)
Permettono il clonaggio di
lunghi frammenti di DNA
Contengono un
centromero, 2 telomeri ed
1 ARS
•Efficienza di
trasformazione bassa
•Sequenze rip. instabili
•ChimerismoBacterial Artificial Chromosome (BAC)
Sito di clonaggio
Contengono i geni parA e
parB che mantengono il
Resistenza antibiotico
numero di copie del
fattore F di E.coli a 1-2
per cellula
Vengono trasferiti in
Origine di replicazione
cellule di coli per
elettroporazione
Inserti di circa 150-250 kb
Vettori basati su repliconi a basso numero di copie (Fattore F di Coli)Librerie Genomiche •Complessità = numero di cloni indipendenti. Può essere definita in termini di Genomi Equivalenti (GE) GE=1 quando il numero dei cloni indipendenti è pari al rapporto dimensioni del genoma / dimensioni medie degli inserti Ad esempio per una libreria di DNA genomico di dimensioni medie di 150Kb, 1 GE = 3000 Mb/150Kb= 20000 cloni indipendenti Per ottenere la rappresentatività di tutte le sequenze di un genoma, si cerca di avere librerie corrispondenti a più Genomi Equivalenti (5-10 X)
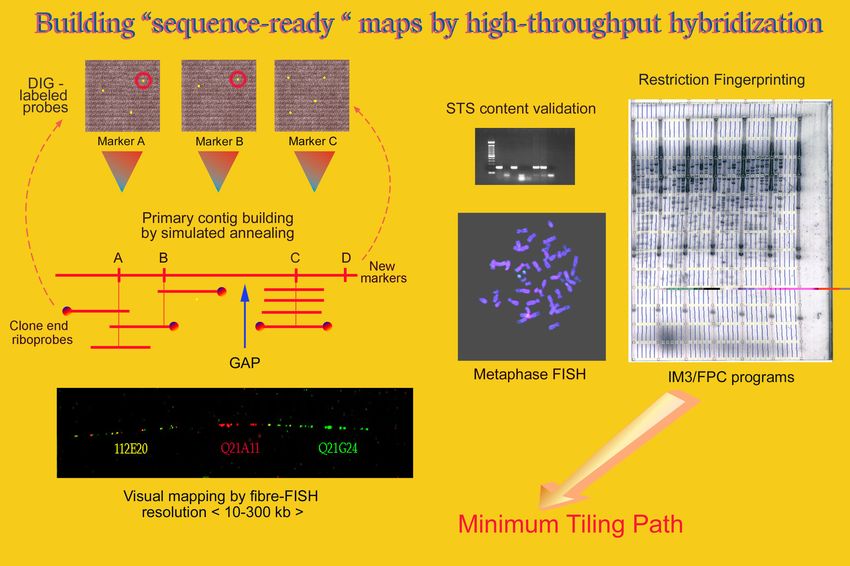
Costruzione della mappa fisica di una regione •Obiettivo: ordinare lungo il cromosoma cloni di DNA con inserti che si sovrappongono - CONTIGUI -. •Le sequenze dei singoli cloni devono essere riconoscibili.
MAPPAGGIO FISICO
Sono possibili diversi livelli di caratterizzazione
MAPPAGGIO A BASSA RISOLUZIONE
(individuazione del cromosoma e della banda citogenetica, distanza tra un clone
e il successivo nell’ordine delle Mb):
•Ibridi somatici
•FISH su cromosomi metafasici
MAPPAGGIO AD ALTA RISOLUZIONE
(distanza tra un clone e il successivo nell’ordine delle kb):
•Mappaggio di restrizione (risoluzione di alcune centinaia di kb)
•FISH su fibre di cromatina (risoluzione da 5 a 700 kb)
•Mappatura mediante STS (sequence tagged site), in cui le posizioni di brevi
sequenze sono mappate mediante PCR o per ibridazione di frammenti genomici.
SEQUENZIAMENTO (risoluzione 1 bp)Ibridi Somatici Permettono di assegnare i cloni ai singoli cromosomi
Metodica
Il nucleo con i due assetti
cromosomici completi è
altamente instabile. Si
assiste normalmente alla
perdita di cromosomi umani
in modo casuale; è possibile
favorire il mantenimento di
un determinato cromosoma o
di un particolare frammento
mediante terreno selettivoDisponendo di una batteria di ibridi somatici per tutti i cromosomi umani è
possibile mappare una determinata sequenza mediante PCR o Southern-blot,
assegnando in questo modo la sequenza ad un determinato cromosoma ->
test di concordanza
Clone ibrido GeneX Cromosomi umani
1
2
3
4
5
6
7
8
A
+
+
+
+
+
-
-
-
-
B
-
+
+
-
-
+
+
-
-
C
+
+
-
+
-
+
-
+
-
Svantaggi legati al fatto che le cellule ibride contengono più cromosomi
umani
Il gene X si trova quindi sul cromosoma 3
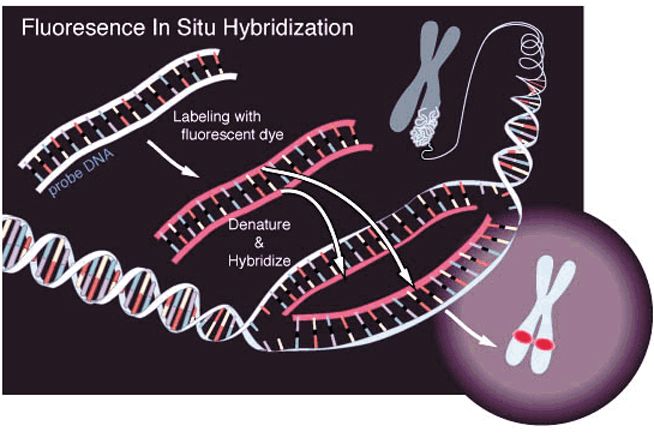
Esistono anche ibridi monocromosomiciL’Ibridazione In Situ con Fluorescenza (FISH) è una tecnica di mappaggio fisico che permette di posizionare i marcatori ibridando una sonda marcata con fluorescenza sui cromosomi intatti, preventivamente fissati su vetrino. L’avvenuta ibridazione è visualizzata mediante microscopio a fluorescenza; A seconda della risoluzione voluta i cromosomi possono essere metafasici o interfasici.
FISH ad alta risoluzione
Risol. Finalità Vantaggi Svantaggi
Cr. metafasici > 1Mb Assegnazione cromosomica Mappaggio ad una Ordinamento difficile
Identificazione di regioni specifica banda
omologhe Orientamento cen-
tel
Cr. stirati >200 kb ordinamento Ordinamento cen- No determinnazione
tel distanza
Nuclei interfasici 50-100 kb ordinamento Determinazione Ordinamento cen-tel
della distanza impossibile
Fibre di DNA 5-500 kb ordinamento Accurata Ordinamento cen-tel
determinazione impossibile
della distanza
FISH su fibre di cromatina = Lisi delle cellule
su un vetrino e scivolamento del DNA
Se il DNA viene deproteinizzato = Fiber
FISH•Fingerprint basato sui siti di restrizione
Mappaggio fisico ad alta risoluzione basato sul mappe di restrizione di lunghi
frammenti di DNA, partendo da DNA genomico o DNA clonato in YAC/BAC/
PAC.
Dipende dalla frequenza di taglio
dell’enzima utilizzato
I cloni vengono identificati come
overlappanti se almeno 1/3 dei
frammenti di restrizione sono in
comuneIdentificazione degli overlapping in base alla somiglianza del pattern di restrizione Facilita il processo di assemblaggio
Mappaggio basato sul contenuto di
STS (Sequence Tagged Site )
Una STS è una corta sequenza di DNA che è possibile mettere in
evidenza con un saggio di PCR. In questo modo si può rapidamente
valutare la presenza o l’assenza della sequenza in questione in
qualsiasi campione di DNA.
Le STS non sono necessariamente polimorfiche ma, se lo sono,
possono essere utilizzate come marcatori sia per le mappe genetiche
sia per quelle fisiche, permettendo di collegarle fra loro.
Rappresentano uno strumento molto importante nelle tecniche di
mappaggioGli STS: Sequence Target Site
L’automazione del sequenziamento permette di sequenziare corte
sequenze (300pb) clonate a caso da cui ricavare primers per “screenare” con la
PCR le librerie e costruire mappe fisiche attraverso la creazione di contigui.
A* C H F*
1 B*
D G*
Q 2
mappa fisica:contiguo
DNA genomico A+,B-,C+..
A C B D G H F Q
B+,D+,G+ A-,B+,C+..
Clonaggio
mappa genetica: A, G e F
H+,F+,T-.. F+,T-,Q+..
sono in linkage il loro
ordine e’ F-A-G
Sequenziamento screening
library con
PCR
H F Q A C B D G
STS A,B,C.. I due contigui sono sullo stesso
scelta dei primers x A,B,C.. cromosoma e via cosi....Come si fa lo
“screening” di una
libreria genomica
5X (100.000 cloni)
in PCR?
100.000 cloni in piastre da 99 pozzetti per
un totale di 1000 piastre.
-Pools primari: pools di 10 piastre (990
cloni) --> 100 PCR
Individuazione del pool primario
positivo.
Trovo un pool di 10 piastre posiivo.- Pools secondari: 1) pools di singole piastre (99 cloni) del pool primario positivo --> 10 PCR. Individuazione della piastra positiva. 2) Pools delle colonne (9 colonne A-I) --> 9 PCR. Individuazione della colonna positiva. 3) Pools delle righe (11 righe 1-11) --> 11 PCR. Individuazione della riga positiva. Quindi, con un totale di 130 PCR, ho individuato il clone positivo su 100.000.
La connessione fra mappe Quindi si hanno due tipi di mappe: fisica e genetica. Come unirle? la mappa fisica mi dice che un gruppo di sequenze formano un contiguo su un frammento di cromosoma, ma non mi permette di identificare geni candidati. La mappa genetica me lo permetterebbe perche’ non riguarda specifiche sequenze, ma anche loci di cui non conosco la sequenza. Non posso pero’ studiare il gene candidato perche’ non ho la sequenza corrispondente. La possibilita’ di generare STS e EST polimorfici ha permesso di risolvere il problema
•Dal 1998 al 1999 sono stati sottoposti a fingerprint 300.0000 cloni
BAC -15 GE
•I dati ottenuti sono stati inseriti in database e ordinati in contigui
che integrano tutti i dati di mappaggio esistenti in banca dati
Clone-by-Clone (Human Genome Project)Il fattore determinante per una molecola di DNA è la sua sequenza nucleotidica. Il Sequenziamento dei Genomi Strategia standard per il sequenziamento di piccoli genomi Clone-by-Clone (Human Genome Project)
Sequenziamento
Ogni clone (150-200 kb) mappato, viene
ulteriormente frazionato e
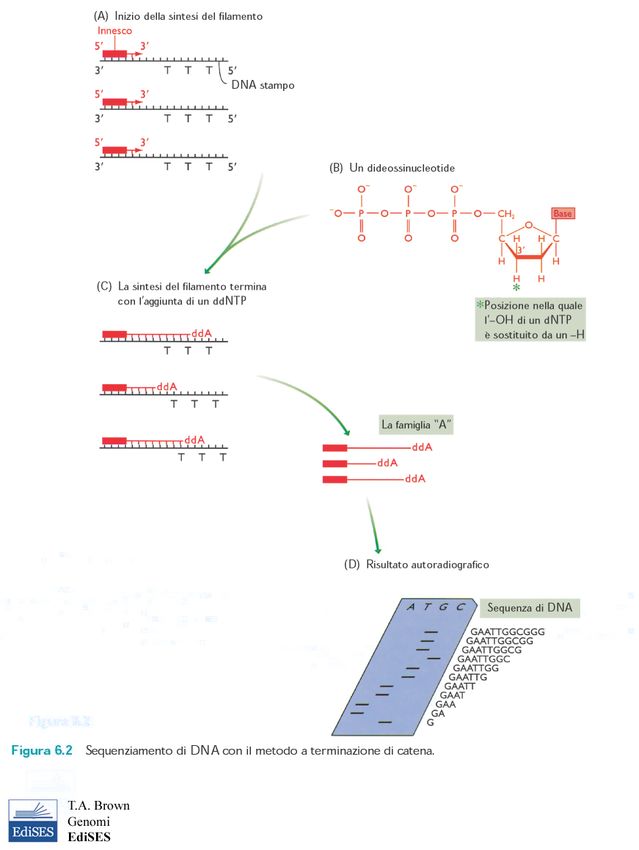
sub-clonato in vettore plasmidicoIl metodo a terminazione della catena
Il sequenziamento automatico del DNA con dideossinucleotidi marcati per fluorescenza
le sequenze dei sub-cloni vengono allineate.....
...fino ad ottenere la sequenza completa del
clone genomico da cui derivavanoGENOMI COMPLESSI Errori dell’analisi dovuti alla presenza di sequenze ripetute
Identificazione di sequenze trascritte Identificare all’interno di contigui genomici la posizione di geni •Il DNA codificante presenta ORF lunghi; •Il DNA codificante è conservato nel corso dell’evoluzione.
1. L’analisi degli ORF
2. Identificazione di isole CpG
Sequenze in genere poste al 5’ dei geni
Il 56% dei geni associato a isole CpG -> tutti i geni housekeeping più il 40%
dei geni ad espressione tessuto specifica
•Identificazione mediante mappaggio di restrizione
Taglio del DNA con
ER rare cutter che
tagliano in sequenze
ricche in GC ->
CGGCCG EagI.
Ibridazione con clone
candidato. La
presenza di siti
ravvicinati indica la
Ibridazione su cloni cosmidici
presenza di un’isola
CpG3. Zoo-blotting
Metodo basato sul concetto
che le sequenze codificanti
sono conservate. Ibridazione
in bassa stringenza del clone
genomico candidato ad una
serie di DNA genomici
estratti da varie specieautosomico Xp/Yp
4. Exon trapping L’identificazione di un gene si basa sulla capacità del clone di produrre splicing in vitro. Si utilizzano vettori in grado di replicarsi in cellule eucariotiche (Ori e prom SV40)
Cellule Cos = hanno integrato un genoma difettivo di SV40 che permette a qualsiasi DNA circolare che contenga a sua volta un’origine di replicazione funzionale di SV40 di replicarsi in modo indipendente dal DNA cellulare.
Mappe di sequenze trascritte
mediante EST
Collocazione di sequenze parziali di cloni di cDNA su mappe fisiche
EST = Expressed Sequence Tags. Sequenziamento a caso
dell’estremità 3’ di cloni di librerie di cDNA -> creazione di primer
specifici per ogni EST da utilizzare per saggi di PCR su cloni di DNA
genomico.Risoluzioni
Tipo di mappa Esempi/metodologia Risoluzioni
Citogenetica Bandeggio cromosomico e Parecchie Mb (5-10 Mb)
FISH; ibridi somatici
Mappe di restrizione Mappe di restrizione con Qualche centinaia di Kb
enzimi rare-cutter
Mappe di contigui di Overlapping di cloni BAC Circa 200 Kb
cloni
Mappe di STS Sono necessarie Meno di 1 Kb
informazioni di sequenza
per ordinare le STS
Mappa di EST E’ necessario sequenziare Circa 90 Kb
cloni di cDNA e ordinarli su
mappe fisiche
Sequenziamento 1 coppia di basiClone contig covering whole chr. 21!
Clonaggio dei geni patologici
Malattia
Malattia
Posizione Posizione
di mappa Funzione Funzione
di mappa
Gene
Gene
CLONAGGIO CLONAGGIO
FUNZIONALE POSIZIONALECLONAGGIO FUNZIONALE
Schema utilizzato per il
clonaggio del Fattore VIII
Isolamento del prodotto proteico corrispondente -> sequenziamento amminoacidico ->
ricerca delle regioni con minima degenerazione dei codoni -> produzione di
combinazioni di oligonucleotidi con tutte le possibili permutazioni -> screening di
librerie cDNA -> screening di librerie genomichesh2/sh2 wild type
X
Topo shaker-2 (sordo) Incroci permettono di mappare sh2 Isolamento dei cloni
sh2/sh2 in una regione di 1cM sul cr. 11 BAC murini contenuti
murino -> regione sintenica a nella regione
17p11.2, candidata per il gene della candidata
sordità DFNB3
Iniezione del clone
in uova sh2/sh2 per
produrre topi
transgenic
BAC 452
corregge il
Isolamento del gene difetto sh2
umano MYO15
Mutazioni a carico Sequenziamento
Mutazione C674Y
di MYO15 identificata nei del BAC 425 ed
identificate in 3 analisi
topi sh2
pazienti DFNB3
Mappaggio di MYO15 computerizzata
in 17p11.2 -> gene myo15POSITIONAL CLONING
Genetic disease
Map to a chromosome site
Retrieve genomic clones that cover the mapped region
Identify and analyze exons
Isolate a cDNA
Isolate a genomic clone
Characterize the normal gene
Mutation detection assaysCLONAGGIO POSIZIONALE Identificazione del gene patologico conoscendo solo la localizzazione cromosomica
•Presenza di un’anomalia citogenetica in un paziente con fenotipo patologico Se l’anomalia citogenetica è localizzata all’interno di un gene patologico già mappato, questo è un buon indice che l’anomalia citogenetica sia causa della malattia •Sindromi da geni contigui -> in questo caso il paziente presenta diverse patologie genetiche in contemporanea •Presenza di ritardo mentale oltre al fenotipo patologico -> sindrome da geni contigui
Identificazione di geni mediante analisi
computerizzata
•Ricerca di omologie in database di sequenze codificanti mediante
l’algoritmo BLAST (basal local aligment sequence tool)
Geni ortologhi: geni omologhi presenti in organismi diversi;
Geni paraloghi: geni omologhi presenti nello stesso organismo,
spesso membri di una famiglia multigenica.•Identificazione di esoni all’interno di una sequenza genomica mediante la ricerca di sequenze conservate tra le giunzioni esoni/introne, siti di biforcazione per lo splicing (Genescan, Grail, Hhmgene, Fex ecc.) •Identificazione di geni attraverso l’uso di pacchetti di software che utilizzano i programmi presenti nei database per la ricerca di omologie, ed i programmi progettati per individuare motivi associati a geni ed esoni e produrre i risultati in grafico -> NIX (UK Human Genome mapping Product Resouce Center e Genotator (US Lawrence Berkeley National Laboratory)
Criteri per l’identificazione di un gene malattia
• Presenza di una mutazione
• comparazione di individui normali e affetti
• comparazione di più individui da famiglie normali e affette
• le mutazioni candidate devono essere trovate solo negli individui affetti
• Le mutazioni possono distruggere la funzione genica
• la mutazione individuata deve interferire con il normale flusso
dell’informazione
• deve essere possibile distinguere tra polimorfismi e mutazioni
• queste possono comprendere: grandi delezioni, mutazioni
frameshift, mutazioni di splicing, mutazioni nonsenso (stop codon)
• La funzione genica anomala deve spiegare la patogenesi
• il gene individuato deve essere normalmente espresso nel tessuto target
• l’espressione è analizzata attraverso:
• Northern blotting, RT-PCR, per l’mRNA
•Western blotting, per l’espressione proteica
• la funzione del gene deve essere consistente con le manifestazioni
fisiologiche della malattia (es., CFTR, distrofina)Ulteriori criteri per l’identificazione
• Correlazione genotipo-fenotipo
• mutazioni diverse correlano con gradi di severità differenti della malattia
• viene fatta la comparazione tra famiglie diverse
• La correzione del difetto “cura” la patologia
• correzione genotipica:
necessità di un protocollo di “gene therapy”
• correzione fenotipica•Screening delle mutazioni •Restaurazione del fenotipo normale per le malattie da loss of function •Produzione del modello murino di malattia Problemi legati allo screening delle mutazioni •Eterogeneità genetica -> importante la selezione dei pazienti secondo criteri clinici molto rigorosi •Omogeneità delle mutazioni •Mutazioni difficili da individuare
The Human Genome Project
Il U.S. Human Genome Project inizia formalmente nel 1990 sotto il coordinamento del U.S. Department of Energy e del National Institutes of Health (NIH).. Gli obiettivi principali del progetto erano: •Costruire una mappa completa del genoma umano •Immagazzinare i dati ottenuti in banche dati •Migliorare gli strumenti per l’analisi delle sequenze •La sequenza completa del genoma umano e di genomi di organismi modello •Trasferire i dati e le tecnologie sviluppate al settore privato •Dare una regolamentazione legislativa ed etica •La previsione era di terminare il progetto in 15 anni ma lo sviluppo tecnologico ha, di fatto, accelerato le tappe
Febbraio 2001
Comparison of methods
Number of human genes
Before genome sequencing After genome sequencing
50.000-200.000 25.000-35.000Full Genome Sequences of
Model Organisms
•E. coli e altri procarioti;
•Saccharomices cerevisiae, utile per le analisi genetiche
(alta frequenza di ricombinazione non omologa);
•Cenorhabditis elegans, utile negli studi dello sviluppo;
•Drosophila melanogaster;
•Topo;
•PrimatiThe Power
of
Comparative GenomicsMouse - Human
overlay
• Overlay mouse fragments on human scaffolds.
• Identify genes and conserved regulatory regions
• Use synteny to aid annotation
• Use additional data to build mouse scaffoldsComparative Genome Sizes of Humans
and Other Organisms Studied
Organism Estimated size Estimated
number of genes
H. Sapiens 3000 million bases 30,000
Drosophila (fruit fly) 180 million bases 13,061
Arabidopsis (plant) 100 million bases 25,000
C. elegans (roundworm) 97 million bases 19,099
S. cerevisiae (yeast) 12.1 million bases 6,034
E. coli (bacteria) 4.67 million bases 3,237 !
Genome size does not correlate with evolutionary status, nor is
the number of genes proportionate with genome size!Ad oggi sono noti circa 6.000 caratteri di tipo mendeliano, descritti nella Banca Dati OMIM. OMIM ® - Online Mendelian Inheritance in Man ®
Puoi anche leggere

















































