Vivere con una malattia rara - Dalla diagnosi alla presa in carico Nuovi strumenti diagnostici - Regione Lazio
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Vivere con una malattia rara
Dalla diagnosi alla presa in carico
18 maggio 2016
Nuovi strumenti diagnostici
Paola Grammatico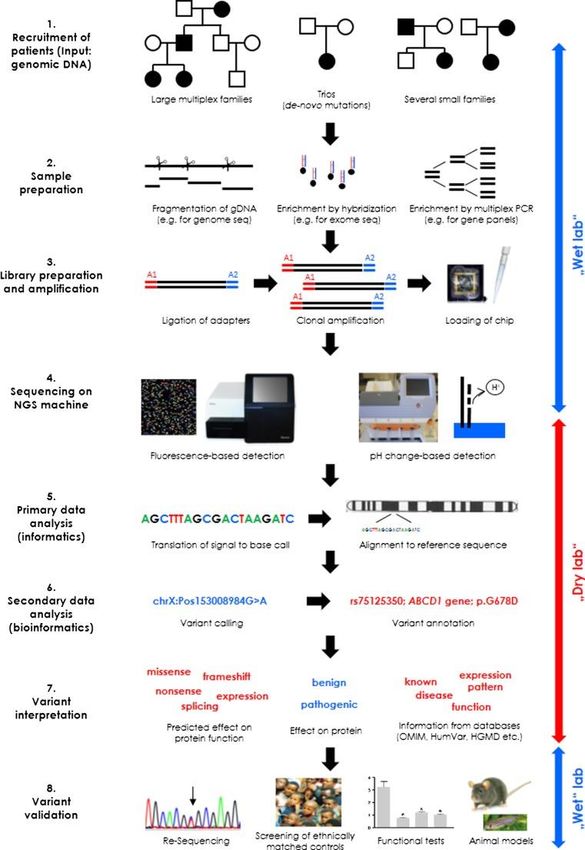
Il Laboratorio di Genetica medica nella
diagnosi delle malattie rare
Citogenetica classica
Citogenetica molecolare
Array-CGH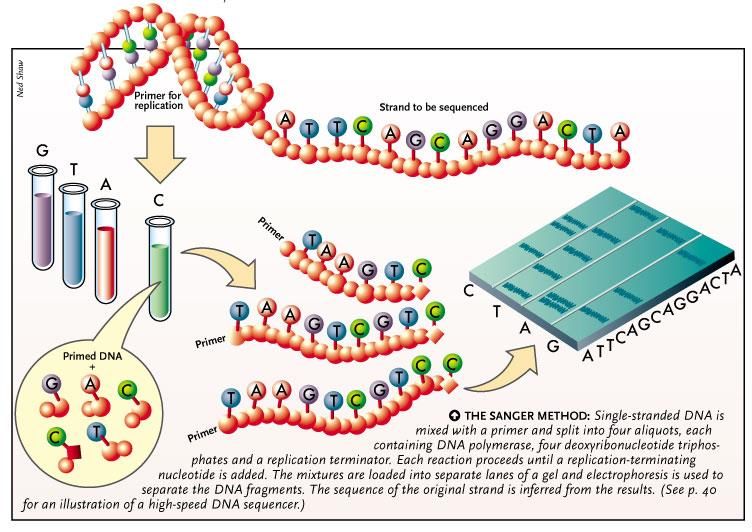
Il Laboratorio di Genetica medica nella diagnosi
delle malattie rare
Reverse dot blot
MLPA
Real time PCR
Sequenziamento: Metodo Sanger
Frederick Sanger: due premi Nobel in Chimica
(1958 e 1980)
Sequenziamento del DNA: Metodo Sanger
1987: primo sequenziatore automatico (Applied Biosystem ABI370)
500.000 pb/giorno output: 500 kb/die
Lunghezza massima del frammento: 600 basi
Analisi di un singolo frammento
amplificato mediante PCR
Gene grande > numerosi frammenti di
PCR > numerose sequenze
La sequenza viene allineata al
riferimento e analizzata per identificare
eventuali varianti nucleotidiche
SEQUENCE REVOLUTIONS
NGS: sequenziamento massivo parallelo
THROUGHPUT
whole genome (WGS)
whole transcriptome NGS
Gb
whole exome (WES)
Più geni / pazienti
in un singolo
esperimento
Throughput
Mb
target seq
Kb
50 300 700
Lenght of Read (bp)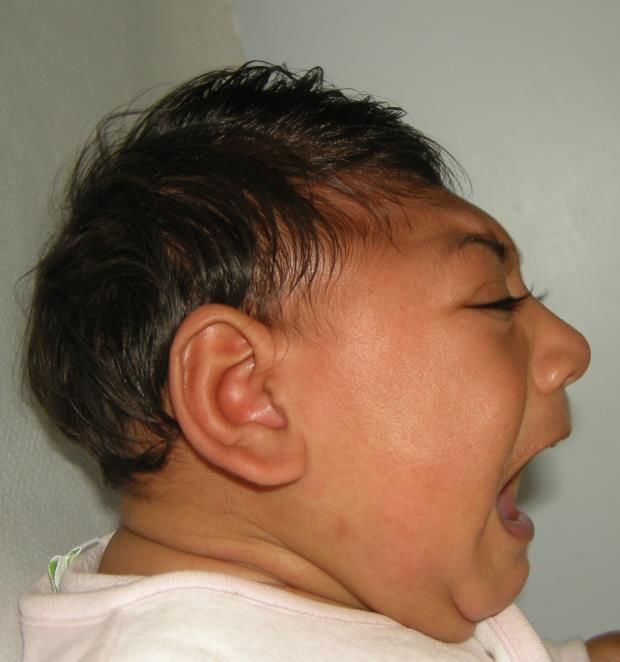
Il Laboratorio di Genetica medica nella diagnosi
delle malattie rare
Ieri…
Oggi..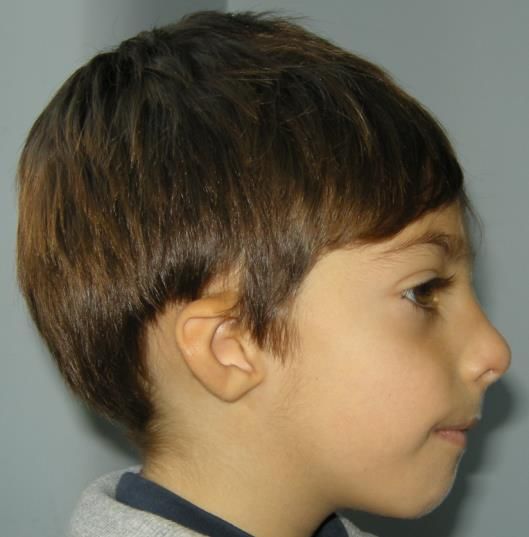
Next generation sequencing (NGS)
Sequenziamento di centinaia di Mb (milioni di bp)
in un’unica seduta analitica
Ricerca: contributo alla identificazione di
nuovi geni responsabili di tratti fenotipici
Diagnosi: incremento delle potenzialità dei
test genetici nella identificazione della causa
molecolare della patologia
SEQUENZIAMENTO NGS
Piattaforme NGS diverse per concezione e chimica ma accomunate dalla capacità di
generare informazioni di sequenza ad alta produttività
(lettura di decine di migliaia di sequenze in parallelo)
PIATTAFORME HIGH THROUGHPUT
CONCEPITE PER SEQUENZIARE AMPIE
REGIONI GENOMICHE
PIATTAFORME MIDDLE THROUGHPUT
CONCEPITE PER SEQUENZIARE
REGIONI GENOMICHE
DI DIMENSIONI MINORI
La tecnologia NGS consente l’esecuzione di indagini in precedenza
tecnicamente non fattibili o economicamente proibitive
Un genoma umano può
essere sequenziato in poche
settimane ad un costo
paragonabile a quello di
alcuni dei test diagnostici
molecolari convenzionaliNGS: potenzialita’ in ambito diagnostico
Diagnosi molecolare di condizioni
precedentemente caratterizzate solo
fenotipicamente
Diagnosi molecolare di malattie associate a
mutazioni in geni di difficile approccio mediante
sequenziamento Sanger
Diagnosi molecolare di malattie a ereditarietà
multigenicaTipologie dei test NGS
Sequenziamento dell’intero genoma
(Whole genome sequencing, WGS)
Sequenziamento dell’intero esoma
(Whole exome sequencing, WES)
Sequenziamento mirato di pannelli di geni
(Target NGS testing o Panel NGS testing)
Il sequenziamento del DNA di nuova generazione: indicazioni per l’impiego clinico: documento SIGU_NGS, gennaio 2016NGS, workflow di un esperimento
Disegno
esperimento
Preparazione del campione
Sequenziamento
Analisi bioinformatica
Risultati
Validazioneoutput dell’esperimento
Allineamento delle letture ottenute
con sequenza di riferimento
Identificazione di TUTTE le varianti
identificazione delle
varianti patogeneticheVarianti di sequenza Varianti causative Varianti responsabili di fenotipi non collegati al quesito clinico (Incidental findings, IF) Varianti di sequenza con effetti funzionali e clinici non definiti (variants of uncertain significance, VUS)
CLASSIFICAZIONE DELLE VARIANTI IDENTIFICATE
American College of Medical Geneticists
Disease causing già descritte come causa della malattia (es.
delezione F508 in CFTR)
Likely disease causing mai descritte in precedenza, ma si suppone
siano causative in base alla tipologia della
mutazione (es. Una mutazione nonsense in
un gene per cui alter mutazioni di questa
tipologia sono state già descritte
Possibly disease causing mai descritte in precedenza; in base al tipo
di mutazione potrebbero o non potrebbero
essere causative di malattia
Likely not disease causing mai descritte in precedenza; probabilmente
non causative di malattia
Not disease causing già descritte come varianti neutrali
Variant of unknown clinical significance varianti nè già descritte nè attese come
causative, ma identificate in associazione a
quadri cliniciNext Generation Sequencing nella diagnosi
delle malattie rare
Whole genome Exome
sequencing sequencing
Target
sequencingFattori determinanti nella scelta dell’approccio • Costi: il costo del sequenziamento di un esoma può oggi essere minore del sequenziamento di un pannello di geni • Finalità: malattie ad elevata eterogeneità genetica/malattie mendeliane (o sospette tali) con gene ancora non noto • Sensibilità: è in funzione del coverage delle sequenze indagate (n. letture “reads” dei singoli tratti del DNA esaminati e del grado di sovrapposizione tra queste) • Probabilità di ottenere IF e VUS: dipende dall’ampiezza della porzione di genoma analizzata e interrogata. • Archiviazione dei dati: necessità di disporre di piattaforme adeguate alla archiviazione di un gran volume di dati.
Sequenziamento dell’intero genoma
(Whole genome sequencing, WGS)
L’analisi mediante WGS non viene ancora
considerata come applicabile a livello
diagnostico per le difficoltà
interpretative.
Il sequenziamento del DNA di nuova generazione: indicazioni per l’impiego clinico: documento SIGU_NGS, gennaio 2016Whole exome sequencing, WES
Sequenziamento delle regioni
codificanti dei geni di un individuo
Indicazioni principali
Patologie genetiche le cui basi molecolari risultino sconosciute
Vantaggi
Analizzare e identificare nuovi geni malattia in pazienti già
sequenziati
Identificare mutazioni in geni malattia noti in fenotipi “atipici”
Limiti
Necessità di un adeguato sistema di archiviazione datiClinical Exome Sequencing
Sequenziamento delle regioni codificanti
dei geni noti per essere associati a
malattia (OMIM)CASO 1
Diagnosi gestaltica
di:
Disostosi
6 anni, caso isolato, genitori non consanguinei
mandibolofacciale,
Alla nascita, fistola tracheoesofagea tipo Guion-Almeida
ad oggi residuano, reflusso
gastro-esofageo ed anemia
Ritardo di acquisizione della parola
Microcefalia (CCCASO 1
Disostosi mandibolofacciale, tipo Guion-Almeida
Disostosi facciale rara
Base molecolare definita nel 2012 (Lines et al., AJHG)
Unico gene: EFTUD2 (no eterogeneità di locus)
EFTUD2
c.[492+4A>G]
Eterozigote, de novo
Delezioni e mutazioni di splicing
comuni in EFTUD2
Nessun laboratorio in Italia Approccio NGS
Tre laboratori in UE TruSight One (Illumina)
(www.orpha.net) (in coll. Dr.ssa Iascone)CASO 2
Diagnosi gestaltica
& radiologica di:
Microcefalia
Nascita/6 mesi, 1° figlio, genitori consanguinei primaria/sindrome
SGA, importante microcefalia alla nascita di Seckel
Fronte sfuggente, overlapping delle suture,
micrograzia
Screening ecografico (addome e cuore) nella
norma
RMN cerebrale: grave microcefalia con
semplificazione delle circonvoluzioniCASO 2
Microcefalia primaria/sindrome di Seckel
Spettro clinico raro con eterogeneità clinica e di locus
Almeno 16 geni fin ora identificati
Tre di questi sono stati identificati solo in s. di Seckel
ASPM
p.[Gln509ter]
Omozigote
Nessun laboratorio in Italia Approccio NGS
Vari laboratori in UE TruSight One (Illumina)
(www.orpha.net) (in coll. Dr.ssa Iascone)Targeted sequencing Indicazioni principali: patologie dove mutazioni in geni noti sono responsabili della maggior parte dei casi (es. Teleangectasia Emorragica Ereditaria – mutazioni geni ENG, ALK1 e SMAD4 nell’80%-87% dei casi) Vantaggi Arricchimento specifiche regioni genomiche Abbattimento dei costi Analisi di un numero elevato di pazienti contemporaneamente Limiti Necessità di un aggiornamento del pannello con l’identificazione di nuovi geni associati alla malattia di interesse
Studio di 62 geni sarcomerici e non sarcomerici in 44 paz con cardiomiopatia ipertrofica mediante NGS Definizione di un protocollo bioinformatico per l’interpretazione delle varianti identificate mediante NGS, sia in ambito di ricerca che in ambito diagnostico
RISULTATI
FILTERING
18.810
(MAFRISULTATI
identificazione di pazienti affetti da cardiomiopatia ipetrofica secondaria
Paziente 10, ♀, 65 anni
MUTAZIONE: GLA c.A644G,p.N215S (eterozigosi)
GLA: malattia di Fabry, X-linked recessiva (1-5/10.000)
Sebbene la malattia di Fabry sia caratterizzata da segni neurologici,
angiocheratoma, insufficienza renale, cardiomiopatia, la pz.10 mostrava solo
cadiomiopatia
Paziente 24, ♀, 7 anni
sindrome di Cantù (MALATTIE DEL SISTEMA GENITO-URINARIO
RENE
POLICISTICO
RENE RECESSIVO
POLICISTICO ARPKD
DELL’ADULTO
ADPKD
Sindrome
SINDROME DI
Branchio-Oto-Renale
MECKEL
(BOR)
SINDROME
MALATTIA OROFACIODIGITALE
VON HIPPEL LINDAU TIPO 1MALATTIE DEL SISTEMA GENITO-URINARIO
RENE POLICISTICO
DELL’ADULTO
ADPKD
RENE POLICISTICO
PKD1, PKD2 RECESSIVO
ARPKD
SINDROME
OROFACIODIGITALE SINDROME DI PKHD1
TIPO 1 MECKEL Sindrome
OFD1 MKS1, MKS2, MKS3 Branchio-Oto-Renale
(BOR)
MALATTIA EYA1, SIX5, SIX1
VON HIPPEL LINDAU
VHL
Analisi di geni selezionati
PANNELLI GENICI SEMPLICIMALATTIE DEL SISTEMA GENITO-URINARIO
SINDROME DI
ALPORT
SINDROME DI
MECKEL
SINDROME DI
BARDET-BIEDL
SINDROME DI
BARTTER
SINDROME DI
JOUBERT
Congenital Anomalies of the
Kidney and the Urinary Tract
(CAKUT)MALATTIE MULTIGENICHE AD EREDITARIETA’ COMPLESSA
SINDROME DI SINDROME DI
ALPORT JOUBERT
AHI1, ARL13B, B9D1, B9D2, C5orf42, CC2D2A, CEP290, CEP41,
COL4A3, COL4A4, COL4A5, MYH9
KIF7, MKS1, NPHP1, NPHP3, OFD1, RPGRIP1L, TCTN1, TCTN2,
TMEM138, TMEM216, TMEM237, TMEM67, TTC21B
SINDROME DI
SINDROME DI
BARDET-BIEDL
BARTTER
ALMS1, ARL6, BBS1, BBS10, BBS12, BBS2,
BBS4, BBS5, BBS7, BBS9, CCDC28B, CEP290, ATP6V1B1, BSND, CA2, CASR,
LZTFL1, MKKS, MKS1, NPHP1, SDCCAG8, CLCNKA, CLCNKB, CLDN16, CLDN19,
TRIM32, TTC8, WDPCP FXYD2, HSD11B2, KCNJ1, KCNJ10,
KLHL3, NR3C2, SCNN1A, SCNN1B,
SCNN1G, SLC12A1, SLC12A3,
SLC4A1, SLC4A4, WNK1, WNK4
Congenital Anomalies of the
Kidney and the Urinary Tract
(CAKUT)
ALDH1A2, BICC1, BMP4, CHD1L, EYA1, FGF20, FOXC1, FRAS1,
FREM1, FREM2, GATA2, GATA3, GDNF, GRIP1, HNF1B, NPHP3,
OSR1, PAX2, RET, ROBO2, SDCCAG8, SIX1, SIX5, SOX17, Analisi di geni selezionati
TFAP2A, UPK3A, WT1
PANNELLI GENICI complessiSINDROME DI BARDET BIEDL
Patologia geneticamente eterogenea (21 geni), trasmessa per lo più come carattere AR; in
alcuni casi è stata osservata una modalità di trasmissione oligogenica
BBS1
Heon et al, 2016
23%
Mutazioni a diversi loci BBS
possono interagire e
determinare o modulare
il fenotipo di alcuni individui
80% dei casi
Mockel et al, 2011NGS: REGOLAMENTAZIONE
Il documento tratta l’ampio range di aspetti etici, legali,
sociali e pratici che scaturiscono dall’uso della
tecnologia NGS in ambito diagnostico-clinico
25 raccomandazioni per implementare l’uso dei test
dagnostici mediante NGS massimizzandone i benefici
e minimizzando i potenziali rischi dovuti ad un loro
impiego non corretto
Definizione criteri standardizzati per la scelta dei geni da inserire nei pannelli
diagnostici, curati, validati e aggiornati da un team multidisciplinare di esperti
(clinici e laboratoristi)• Raccomandazioni operative per i
test NGS
• Consulenza genetica nei test NGS
(Peculiarità e Contenuti specifici)
• Comunicazione dei risultati
(consulenza post-test)
• Analisi dei dati NGS e requisiti di
qualità
• Conservazione dei dati NGSProblematiche rilevanti:
Incidental findings (IF)
Identificazione di varianti a carico di geni responsabili
di patologie non correlate al quadro clinico per cui si
sta eseguendo l’accertamento.
Riscontro che abbia potenziale importanza per la
salute o rappresenti un rischio riproduttivo
Consenso informato
Consulenza geneticaIl Laboratorio di Genetica medica nella diagnosi delle
malattie rare
Diagnosi prenatale non invasiva
Diagnosi preimpiantoNIPT - Non Invasive Prenatal Testing cffDNA: DNA fetale libero circolante Origina da cellule placentari e viene rilasciato nel sangue materno cffDNA è presente nel plasma materno a partire dalla IV-V settimana di gestazione e la sua concentrazione (mediante compresa tra il 2% e il 40%) aumenta proporzionalmente con l’età gestazionale. Entro circa 2 ore dal parto non è più presente nel plasma materno FRAZIONE FETALE (FF): % di cffDNA nel plasma materno. Per poter effettuare NIPT la FF deve essere almeno il 4%
NIPT - Non Invasive Prenatal Testing Il DNA libero circolante viene sequenziato e il numero di sequenze per ogni cromosoma viene confrontato con un genoma di riferimento. Se il numero di sequenze di un cromosoma si discosta dall’atteso, viene segnalata un’aneuploidia Anomalie cromosomiche indagate: trisomia 13, 18, 21 + aneuploidie dei cromosomi sessuali Riduzione attesa del ricorso a procedure di diagnosi prenatale invasiva: 95%
NIPT - Non Invasive Prenatal Testing
FATTORI CHE INFLUENZANO AFFIDABILITA’ DEL TEST
-Mosaicismi confinati alla placenta
-Vanishing twin
-Peso della madre (influenza Frazione Fetale)
-Fumo, alcol, droghe da parte della madre (influenzano
Frazione Fetale)
-Infezioni materne
-Neoplasie materne
-Trasfusioni di sangue, trattamento con cellule
staminali, trapianti materni
LA NIPT è un test di screening (non diagnostico)Il Laboratorio di Genetica medica nella diagnosi delle
malattie rare
Diagnosi prenatale non invasiva
Diagnosi preimpiantoCHI RICHIEDE PGD
Chi richiede la PGD
COPPIE CON RISCHIO AUMENTATO PER UNA PATOLOGIA EREDITARIA GRAVE
• Coppie a rischio infertili
• Coppie fertili con rischio 25-50%
• Coppie a rischio con una storia di ripetuti aborti per
feto affetto e/o che rifiutano abortoNuovi strumenti diagnostici nel
Laboratorio di Genetica medica
Conclusioni
Implementazione delle capacità diagnostiche
dei test genetici
Standardizzazione dei percorsi di validazione
dei test genetici
Centralizzazione dei test genetici di II livello
(DCA 549 del 18-11-2015)Nuovi strumenti diagnostici nel
Laboratorio di Genetica medica
Conclusioni
Adeguamento delle dotazioni organiche
dei Laboratori di Genetica medica
Implementazione degli ambulatori
di consulenza genetica
Forte interazione tra genetisti di laboratorio
e genetisti clinici
Nuovo nomenclatore/tariffarioPuoi anche leggere

















































