Le ultime novità sul database europeo EUDAMED e il codice di identificazione del dispositivo UDI - Assobiomedica
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Le ultime novità sul database europeo EUDAMED e il codice
di identificazione del dispositivo UDI
discusse durante l’incontro del gruppo di lavoro composto dalla
Commissione (COM) gli stati membri (SM) e alcune associazioni europee,
a Bruxelles il 4 maggio
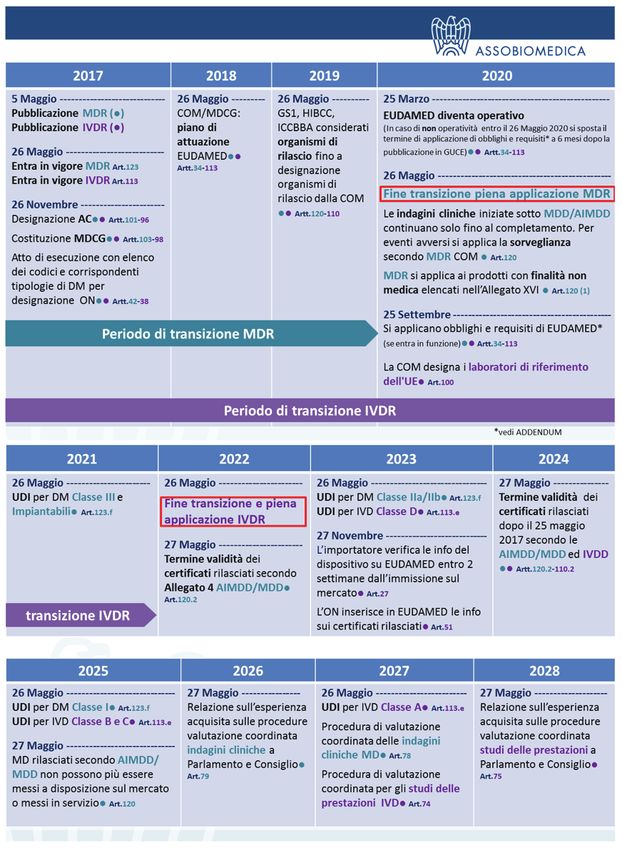
Il rapporto della Commissione chiarisce i requisiti dell’UDI, di Eudamed e le date di applicazione in
riferimento ai due Regolamenti. Il codice di identificazione UDI dovrà essere riportato sui dispositivi in
base alla classe di rischio entro le date prestabilite nei regolamenti ai seguenti articoli:
• MDR - Art. 123 (f).
• IVDR - Art. 113 (e).
I dispositivi riutilizzabili devono riportare l’UDI sul dispositivo stesso entro 2 anni dalla data prevista
per la rispettiva classe di rischio.
Il Medical device coordination group (MDCG), di cui fanno parte 2 esperti per Stato membro (1 per
DM, 1 per gli IVD) è essenziale per l’attuazione del database. La sua istituzione è prevista entro il 26
novembre 2017.
La COM designerà tramite atti di esecuzione gli Organismi di rilascio per l’assegnazione dell’UDI,
che in attesa resteranno i codici: GS1, HIBCC, ICCBBA
La definizione di UDI-DI di base in Eudamed:
• Unico UDI-DI base per dispositivo (un codice per singolo dispositivo);
• UDI-DI di base deve avere di i dati del tipo di dispositivo;
• Possibilità di avere più UDI-DI per lo stesso dispositivo i quali sono associati ad un unico UDI-DI di
base;
• Possibilità di associare l’unità di utilizzo del DI per imballaggio di base (livello 1) per l’UDI-DI (che
possono essere molteplici per un UDI-DI di base);
Aspetti discussi nel corso dell’incontro:
a) Problemi derivanti dalle disposizioni del MDR sulla possibilità di registrare dispositivi di classe I
prima che la banca dati Eudamed sia operativa. Fino a che il database Eudamed non è pienamen-
te operativo, sarà prevista una deroga in quanto è impossibile per un dispositivo essere conforme
agli obblighi dei Regolamenti senza il Single registration number (che verrà assegnato solo dopo
che Eudamed è attivato).
b) I regolamenti prescrivono che la COM, in collaborazione con l’MDCG, elabori le specifiche fun-
zionali di Eudamed. La COM provvederà a:
»» emettere un piano per l’attuazione di tali specifiche funzionali per Maggio 2018, mentre i con-
trolli avverranno dopo Agosto 2019 affinché Eudamed sia pienamente operativo entro Marzo
2020 dopo la pubblicazione in GUCE del relativo annuncio.
»» Presentare i principi inerenti la registrazione degli attori e la gestione degli utenti.
c) Qualora Eudamed non fosse funzionante entro il 26 maggio 2020 per cause non prevedibili, i
relativi obblighi e prescrizioni si applicano entro sei mesi dalla pubblicazione dell’avviso in GUCE
della funzionalità di EUDAMAED (25 marzo 2020). Tra gli obblighi:
1
1) registrazione dei dispositivi;
2) registrazione degli operatori economici;
3) summary of safety clinical performance (SSCP) per i dispositivi ad alto rischio;
4) domande di autorizzazione di indagini cliniche da parte dello sponsor;
5) registrare e notificare eventi avversi nel corso delle indagini cliniche;
6) periodic Safety update report;
7) notifica di incidenti, FSCA e FSN;
8) trend report.
d) La Commissione ha indicato che il termine attore definisce:
»» un’organizzazione con un ruolo e rispettive gerarchie: fabbricanti, mandatari, importatori e di-
stributori. Ogni attore avrà il suo SRN, non sarà unico per organizzazione, per cui un’azienda
si potrà registrare più volte in quanto avrà più attori in Eudamed con i rispettivi SRN;
»» il mandatario dovrà essere registrato ed avere il relativo SRN prima che il fabbricante extra -EU,
da lui rappresentato, si possa registrare.
I principi di gestione degli utenti sono incentrati sul fatto che un:
–– local user administrator (LUA) ottiene l’accesso a Eudamed insieme al proprio attore respon-
sabile per la registrazione;
–– l’accesso del LUA ed il relativo SRN diventerà attivo una volta che la registrazione dell’attore
è validata dall’Autorità Competente;
–– il LUA è responsabile per la gestione delle richieste di accesso da parte di altri utenti associati
all’attore su Eudamed;
–– la Commissione consentirà l’accesso al primo LUA autorizzato dalla AC;
–– le AC consentiranno l’accesso agli ON e agli operatori economici del primo LUA.
e) Registrazione degli attori con i rispettivi LUA: la COM ha presentato i modelli e le regole per effet-
tuare la registrazione degli attori. Saranno previsti nuovi form standard per la registrazione degli
stessi insieme ad un accordo sulla responsabilità in materia di informazioni che ogni attore dovrà
fornire. Solamente il primo LUA è autorizzato; anche se, costituirà buona prassi per l’attore avere
2 amministratori locali.
»» il primo LUA può trasmettere agli altri utenti i diritti a lui spettanti
»» Ogni utilizzatore accetterà un disclaimer su diritti e obblighi prima di accedere ad Eudamed per
la prima volta in associazione ad uno specifico attore.
f) Doppie registrazioni, la COM ha spiegato che controllerà la presenza di doppie registrazioni degli
attori in Eudamed:
»» all’inizio della registrazione dell’attore;
»» alla presentazione della domanda dell’attore;
»» a livello di AC.
In caso di potenziali duplicati la COM invierà un avviso che non bloccherà la registrazione ma
comporterà l’obbligo per l’attore di fornire apposita giustificazione. Al momento della registrazio-
ne, l’attore avrà la possibilità di fornire documenti ufficiali che giustificano l’accesso ad Eudamed.
2g) Molti dei contenuti in Eudamed saranno pubblici se non contengono dati privati o commerciali
sensibili. La COM fornirà linee guida alle AC sulle procedure di validazione.
h) La COM ha comunicato modelli e regole da seguire per le richieste di accesso e la gestione dei
LUA:
»» il principio generale è che il LUA ha pieno governo dei suoi attori utenti;
»» il LUA non può creare account in anticipo; l’utente deve fare richiesta di accesso;
»» importante che l’utente abbia un accordo per essere registrato in Eudamed;
»» l’AC che ha validato un attore sarà informato qualora cambi il LUA.
Nei prossimi incontri del WG sarà prodotto un documento con i profili dei diversi utenti e la descri-
zione delle azioni che possono essere espletate su Eudamed.
i) Mandatario:
il mandato è il contratto alla base tra il mandatario e il fabbricante extra-EU che entrambi devono
sottoscrivere.
»» Il fabbricante rimane responsabile per la registrazione dei dati dei suoi attori in Eudamed ed il
mandatario partecipa alla loro validazione,
»» il fabbricante deve fornire al mandatario i dati del mandato,
»» Il mandatario controlla le richieste dei fabbricanti extra-EU e le inoltrerà alle AC attestandone la
loro conformità.
»» L’AC informa la COM in caso di rigetto delle richiesta da parte di un fabbricante extra-EU per
frode. Se il rigetto è dovuto per mancanza di informazioni o di informazioni inaccurate il form
sarà rispedito all’attore che registra per essere completato.
j) Nel caso in cui due persone dello stesso attore provino a registrarsi ognuno avrà un proprio EU
Login ID.
Conclusioni
Le novità esposte dalla Commissione, un elaborazione dei lavori dei WG di Eudamed e UDI a cui
stanno lavorando per fornire maggiori chiarimenti, restano comunque ancora poco chiare per la ap-
plicazione dei requisiti. Le attività, nel momento in cui dovranno essere eseguite, risulteranno comples-
se e richiederanno ulteriori chiarimenti per comprendere la loro applicazione ottimale. Le disposizioni
sinora analizzate confermano la nostra percezione in termini di:
• alti investimenti economici,
• burocrazia e allungamento dei tempi,
• [molte risorse da dedicare,
• maggiori strategie e processi per l’adeguamento.
Nel momento in cui saremo in grado di fare quanto sopra esposto, sicuramente non avremo uno snelli-
mento del lavoro e tantomeno una semplicità nello svolgimento dello stesso poiché i requisiti sono tanti
e non chiari nella loro applicabilità. Sapremo di più a fine Settembre al prossimo incontro dei WGs
su questi due significativi temi.
34
5
Puoi anche leggere

















































