E' possibile una diagnosi precoce? - Approccio multidisciplinare alle malattie neuromuscolari - MCA Scientific Events
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù

Approccio multidisciplinare alle malattie
neuromuscolari
E’ possibile una diagnosi precoce?
Neuropediatric
18 Ottobre 2017
Drssa Isabella Moroni
Divisione di Neuropsichiatria Infantile
Fondazione IRCCS Istituto Neurologico C.Besta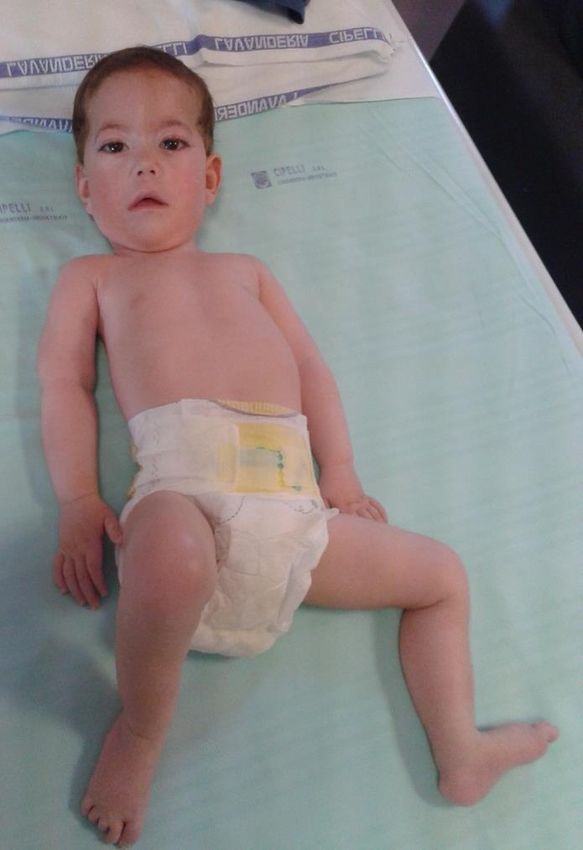
MALATTIE NEUROMUSCOLARI
Importanza diagnosi precoce
1. Counselling familiare decisioni informate della
coppia su successive gravidanze
2. Accesso a presa in carico adeguata trattamento
riabilitativo, monitoraggio complicanze
(respiratorie, cardiomiopatia, ortopediche)
3. Avvio a terapie per forme specifiche beneficio
maggiore se patologia muscolo è iniziale = età
precoce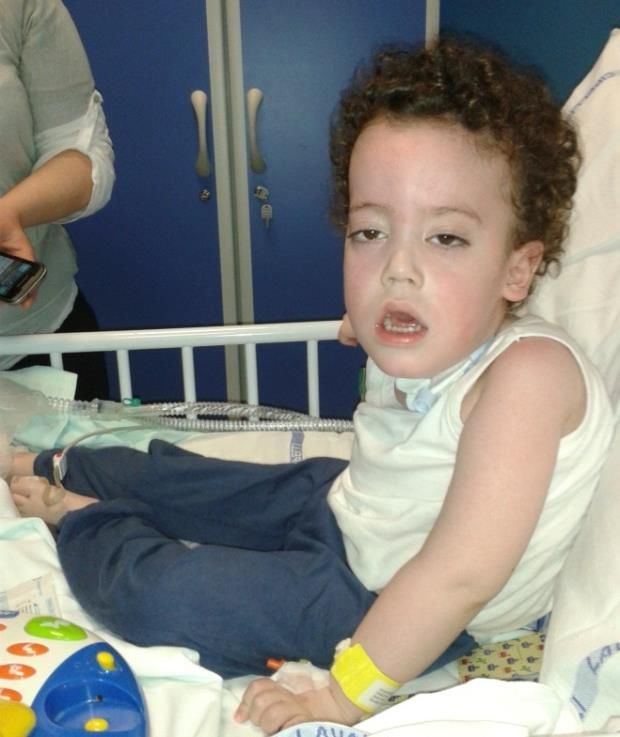
FORME GENETICHE
• DISTROFIE MUSCOLARI PROGRESSIVE
Distrofinopatie
Distrofie dei cingoli
Facio-scapolo-omerale
• ATROFIA MUSCOLARE SPINALE (SMA)
• DISTROFIE MUSCOLARI E MIOPATIE
CONGENITE
• MIOPATIE METABOLICHE
FORME IMMUNOMEDIATE
• MIASTENIA GRAVIS
• DERMATOMIOSITE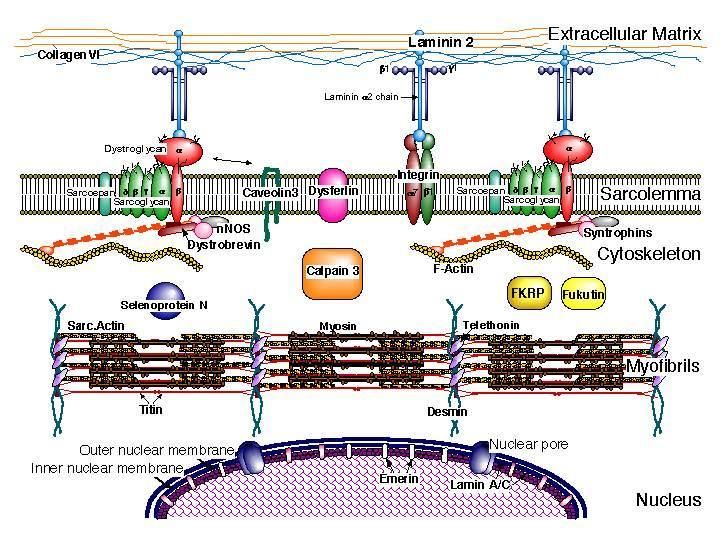
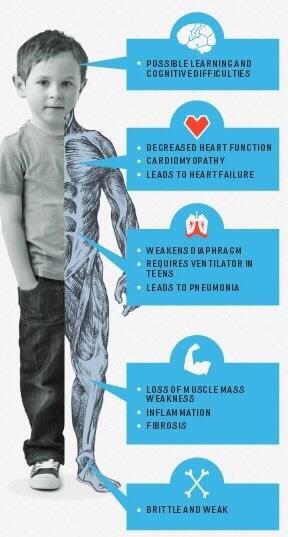
DISTROFIA DI DUCHENNE Frequenza = 1:5000 maschi • esordio 2-5 anni ipostenia prossimale arti inferiori - difficoltà nel rialzarsi da terra (segno di Gowers) - alterazione del cammino - tendenza ad appoggio su punte - difficoltà nella corsa o scale - ipertrofia polpacci • frequente lieve ritardo cognitivo, soprattutto difficoltà • CPK molto elevate 50-100 nell’area del linguaggio volte il massimo • Spesso riscontro «occasionale» di iperCKemia >> 1000 • E anche transaminasi… non segno disfunzione epatica!
Evoluzione • coinvolgimento prossimale arti superiori • progressiva ipostenia arti inferiori perdita cammino autonomo dopo i 10-13 anni • Cardiomiopatia 25% a 6 anni 59% a 10 anni • Retrazioni articolari multiple • Scoliosi deformità toracica Exitus per coinvolgimento respiratorio e cardiaco
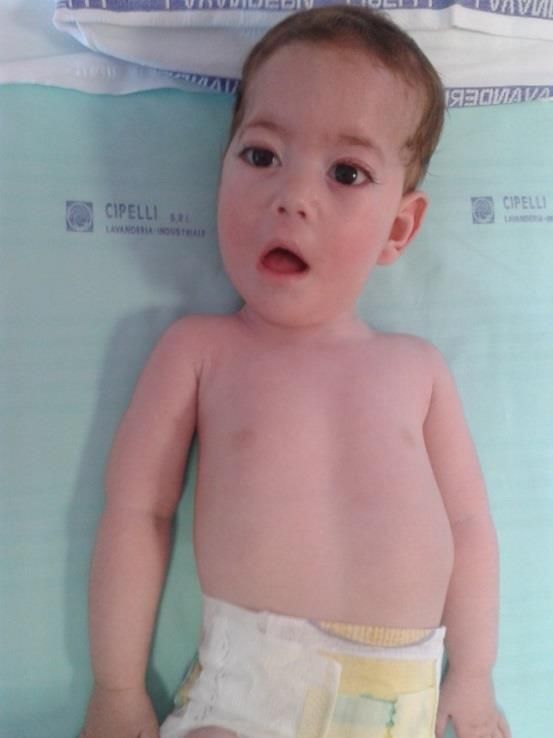
DISTROFIA DI BECKER • Incidenza 1:20000 maschi • esordio molto variabile, 5-15 anni, anche età adulta • ipostenia negli stessi distretti, meno grave • > ipertrofia polpacci • CPK meno elevate • cardiomiopatia tardiva • evoluzione meno grave e più lenta •Dotazione cognitiva normale, rare segnalazioni di compromissione QI
DISTROFINA E GENE DYS
Trasmissione X-linked recessiva
Distrophyn
XY X
X
X X X X
X X Y Y
omozigote
eterozigote
eterozigoteaffetto
sano sano sano
Controllo
normale >> delezioni 25% 50% 25%
mut puntiformi
DMD
duplicazioni
DEFINIZIONE MUTAZIONE
BMD
=
POSSIBILE DIAGNOSI PRENATALE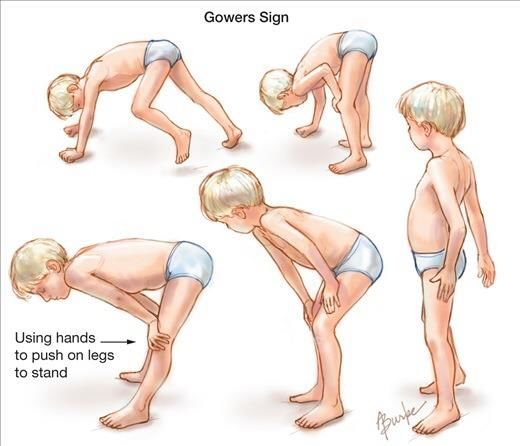
Domanda 1. La diagnosi precoce è utile per: 1. Consulenza di coppia 2. Trattamento riabilitativo, avvio a terapie specifiche, monitoraggio complicanze 3. Orientamento scolastico e sportivo 4. 1 e 2
MOTIVI DI RITARDO DIAGNOSI - Sintomi variabili e aspecifici nei primi anni - Primi sospetti in famiglia non specifici e sottovalutati ritardo cammino e linguaggio - Prima richiesta di valutazione a medico base non esperto in pediatria o patologia muscolare - Frequente riferimento a specialista Ortopedico
156 DMD e familiarità negativa (dal 1982 a 2000)
RITARDO
DIAGNOSTICO
MEDIO 2.5 yrs
Età media alla diagnosi 5 aa15 centri
384 pz
Età media primo sospetto = 31 m
Aumento CK e transaminasi 53% Specialista centro I liv
Ritardo motorio 16 % Pediatra
Ipostenia 14% Specialista centro III liv
Familiarità positiva 7.8% Genitori
Disabilità cognitiva 2.6%
Ritardo linguaggio 4.1%
Età media diagnosi= 41 m
Valutare livelli CK riduce T diagnosiCOINVOLGIMENTO COGNITIVO NELLA DMD
Difficoltà cognitive e comportamentali si associano in 1/3 dei casi
81 pz < 4 yrs -
Valutazione con Scala Media totale QS 87 (SD 15.3)
Griffith
In 55.5 % QS normale > 85
In 32 % QS borderline 70-84
In 12.3 % QS < 70
correlazione tra difficoltà cognitive e tipo di mutazioniDomanda 2: Quando sospettare Distrofinopatia ? 1. Soggetto Maschio 2. Alterazione nei passaggi posturali e deambulazione +/- associati a difficoltà cognitive > linguaggio 3. Riscontro occasionale di transaminasi elevate e CK > 1000 4. Tutte le risposte
Invio a centro specializzato per
malattie neuromuscolari
DAL SOSPETTO ALLA
DIAGNOSI DEFINITAAtrofia muscolare spinale
• Incidenza 1:6000-10,0000 nati
• Trasmissione AR
• Degenerazione dei motoneuroni del midollo spinale
Alterazione del gene SMN1
codifica per proteina
espressa ubiquitariamente e
in particolare nei
motoneuroni del midollo
spinale
Il numero di copie di SMN2
influenza la gravità del fenotipoSMA I • Esordio < 6 mesi, grave sindrome ipotonico-areflessica, ipostenia prossimale, atteggiamento degli arti inferiori “a rana” pianto e riflesso tosse deboli • Ipostenia > prossimale e maggiore arti inferiori, risparmio muscolatura facciale, atrofia e fascicolazioni lingua • Ipostenia muscolatura intercostale con risparmio di quella diaframmatica respiro paradosso, torace a campana • Dotazione cognitiva normale • Deficit di suzione, deglutizione difficoltà alimentazione • Evoluzione rapida e fatale
SMA II
• Esordio 6-18 mesi acquisizione posizione seduta senza
supporto, successiva mancata acquisizione cammino
autonomo
• Ipotonia, ipo-areflessia, ipostenia prossimale
• Frequenti tremori fini distali agli arti superiori
• Dotazione cognitiva normale
SMA III
• Esordio > 18 mesi, ritardo cammino
• Severità molto eterogenea
• Acquisizione cammino autonomo in età tardo infantile
sviluppo di ipostenia prossimale progressiva sino a perdita
cammino in casi severi
Diagnosi
SOSPETTO • CPK: N
• EMG
• Analisi geneticaDati da 21 pubblicazioni 2010-2014 Ritardo maggiore in SMA II e III Il ritardo è dovuto >> a indirizzo a visite specialistiche varie, non mirate
Domanda 3. Nel bambino con ipotonia: 1. È sempre necessario prescrivere EMG 2. E’ sempre necessario prescrivere RMN encefalo 3. È sempre utile prevedere biopsia muscolare 4. Nessuna delle precedenti
MIOPATIE CONGENITE AD-AR- X linked 1. Esordio precoce: congenito primi mesi 2. Ipotonia e ipostenia > assiale e facciale 3. Spesso si associano segni dismorfici/malformativi: artrogriposi multipla, petto escavato, cifoscoliosi, displasia congenita dell’anca, volto allungato, palato ogivale 4. Possibile coinvolgimento SNC (ritardo mentale o epilessia) 5. Evoluzione stabile o lentamente progressiva, peggiore nelle forme ad esordio più precoce SOSPETTO DIAGNOSI CPK: per lo più N o poco elevate Indispensabile BIOPSIA MUSCOLARE E STUDI GENETICI ESTENSIVI
RYR1 MTM1 TTN
IPOTONIA E IPOSTENIA ASSOCIATI AD ALTERAZIONI SNC
DISTROFIE MUSCOLARI CONGENITE - AR
1) Forme pure
Debolezza muscolare diffusa, assenza/difficoltà controllo
capo e posizione seduta, assenza/difficoltà gravi
cammino
2) Forme con coinvolgimento SNC:
RITARDO INTELLETTIVO
EPILESSIA
ANOMALIE OCULARI
(miopia, glaucoma, cataratta,
retinopatia, atrofia nervo ottico)
DEFICIT UDITO e LINGUAGGIO ALTERAZIONI RM
ENCEFALO (sostanza bianca
e/o corticali e/o cerebellari)
CPK non sempre elevate
BIOPSIA : studi immunoistochimici
STUDI GENETICI ESTENSIVISINTOMI DA NON SOTTOVALUTARE:
AFFATICABILITA’ E IPOSTENIA FLUTTUANTI
Miastenia Gravis
• Esordio dopo i 2 anni
• Forme Oculari – Generalizzate
• Ptosi palpebrale, alterazioni motilità oculare
(diplopia)
• Difficoltà masticazione e disfagìa
• Disfonìa
• Esauribilità dopo sforzo
• Faticabilità generalizzata
• Fluttuazione sintomi, peggiori alla sera
CPK normali
EMG con stimolazione ripetitiva, test singola fibra
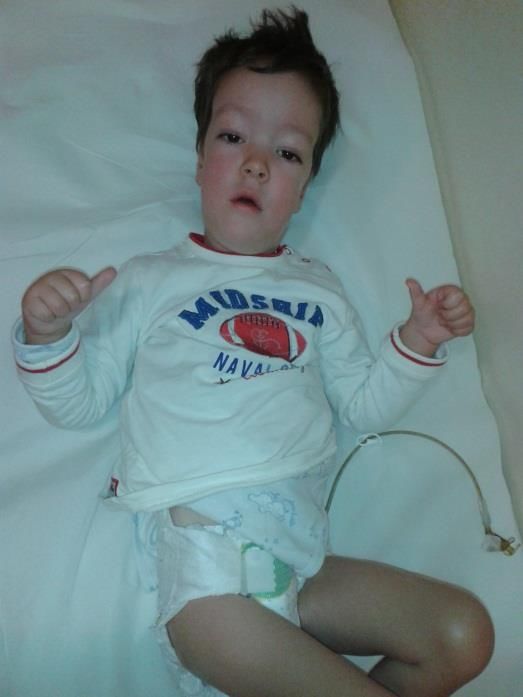
Dosaggio Anticorpi antiAchR, anti MUSK (+ in 90%)MIALGIE E IPOSTENIA (SUB) ACUTA
• Esordio acuto o subacuto Dermatomiosite
• Dolori muscolari diffusi
• Ipostenia > prossimale
• Alterazioni cutanee volto, mani, gomiti, ginocchia (non
sempre marcate)
• Evoluzione con retrazioni grosse articolazioni (gomiti, anche,
ginocchia)
Diagnosi: CPK e indici infiammatori elevati
EMG, biopsia muscolareSOSPETTA PATOLOGIA NEUROMUSCOLARE
• RITARDO MOTORIO +/- RITARDO COGNITIVO considerare età, sesso
• IPOTONIA considerare distretti
• IPOSTENIA considerare se arti, facciale, assiale
se costante o fluttuante
se esordio acuto
• SEGNI e SINTOMI ASSOCIATI dismorfismi, malformazioni, SNC
affaticabilità, mialgìe
• Anamnesi per familiarità definita o sospetta; valutare se consanguineità
• Esecuzione CPK
NEL SOSPETTO inviare rapidamente a
centro di riferimento neuromuscolare
pediatricoDomanda 4. E’ POSSIBILE UNA DIAGNOSI PRECOCE ? 1. Sì sempre 2. No mai 3. Qualche volta 4. È possibile valutando accuratamente segni e sintomi
DALLA DIAGNOSI ALLA PRESA IN CARICO
PEDIATRA
Approccio multidisciplinare
e multicentricoUO Neuropsichiatria Infantile UO Neurologia dello Sviluppo
DIPARTIMENTO NEUROSCIENZE PEDIATRICHE
GRAZIEPuoi anche leggere

















































