DATABASE ITALIANO DELLA SINDROME DI NOONAN E DELLE RASOPATIE - Ripristinato
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Università degli Studi di Torino Città della Salute e della Scienza di Torino
Dipartimento di Scienze della Sanità Pubblica e Pediatriche Ospedale Infantile Regina Margherita
DATABASE ITALIANO DELLA SINDROME DI
NOONAN E DELLE RASOPATIE
Sviluppo del database nazionale per lo studio approfondito delle caratteristiche
cliniche e dell'evoluzione della Sindrome di Noonan e delle Rasopatie
Dott.ssa Diana Carli
25 maggio 2019, Caorle
Assemblea dei soci Associazione Nazionale Sindrome di Noonan e RASopatie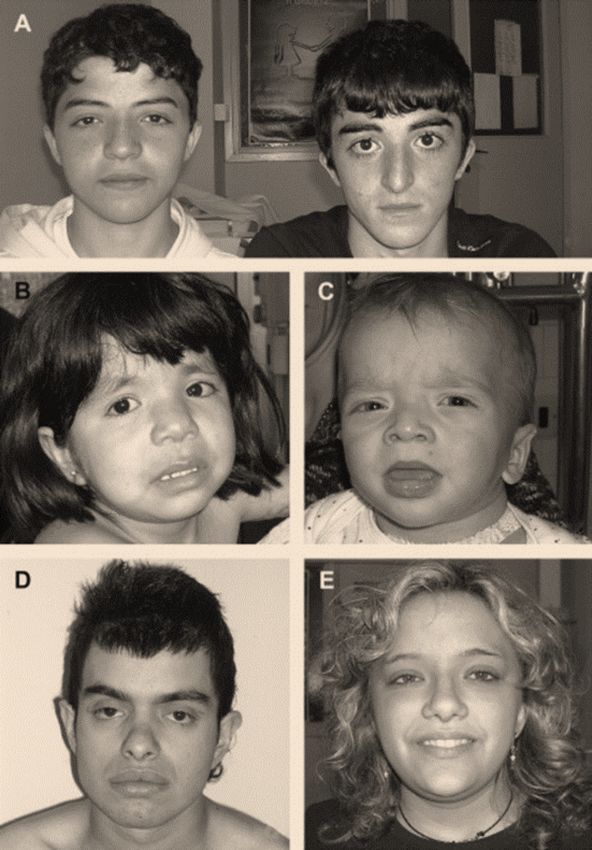
La Sindrome di Noonan Prevalenza
1:1000-1:2500 nati vivi
Cenni Storici
Criteri maggiori
Kobilinsky O. Ueber Eine Flughoutahnbiche Ausbreitung. Am Halse Arch Facies tipica
Antropol 1883; 14:342-8 Stenosi/displasia valvola polmonare
Maschio 20 anni con collo corto e facies suggestiva
o cardiomiopatia ipertrofica
Ullrich O. Ueber Typische Kombinationbilder Multiper Abanturgen. Z Statura < 3° centile
Pectus carenatum/excavatum
Kinderheilkd 1930; 49:271-6
Ritardo psicomotorio +
Pazienti di entrambi i sessi con collo corto e bassa statura criptorchidismo + displasia
linfatica
Turner HH. A syndrome of infantilism, congenital webbed neck, and cubitus (Parente di primo grado affetto)
valgus. Endocrinology 1938; 25:566-74
Criteri minori
Donne con infantilismo sessuale, collo corto e bassa statura Facies suggestiva
Altra cardiopatia congenita
Ford C, Jones K, Polani P, Bruiggs B. A sex chromosome anomaly in a case of Statura < 10° centile
gonadal dysgenesis (Turner’s syndrome). Lancet 1959; I:711-3 Torace allargato
Sindrome di Ullrich-Turner Ritardo psicomotorio o
criptorchidismo o displasia
linfatica
Noonan JA, Ehmke DA. Associated noncardiac malformations in children (Parente di primo grado suggestivo)
with congenital heart disease. Midwest Soc Pediatr Res 1963; 63: 468–70
Diagnosi Clinica
Opitz J. Editrorial comment: The Noonan syndrome. Am J Med Genet 1985; Facies tipica + 1 criterio maggiore o
21:515-18 2 minori oppure
Sindrome di Noonan Facies suggestiva + 2 criteri maggiori
o 3 minori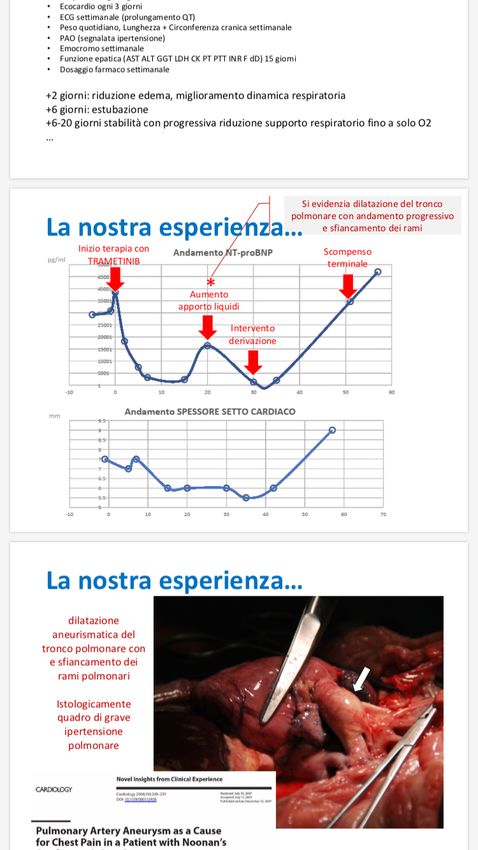
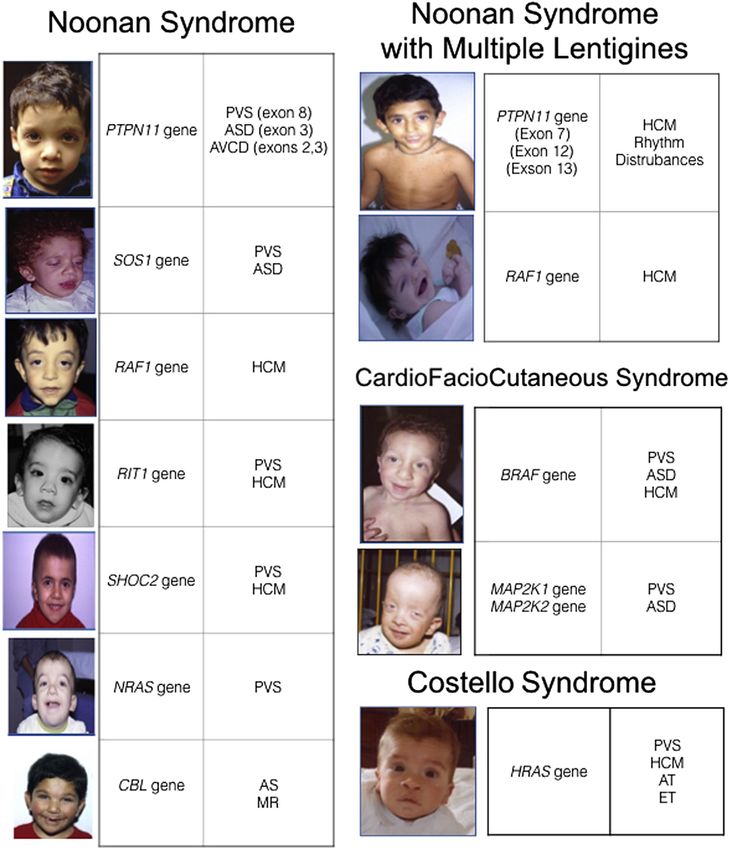
La Sindrome di Noonan
Facies Anomalie scheletriche
Le Rasopatie
Sindrome Cardiofaciocutanea Sindrome di Noonan con
Sindrome di Costello
lentigginosi multipla (LEOPARD)
From Gripp et al. 2011 From Pierpont et al. 2014
Neurofibromatosi di tipo 1
Sindrome Legius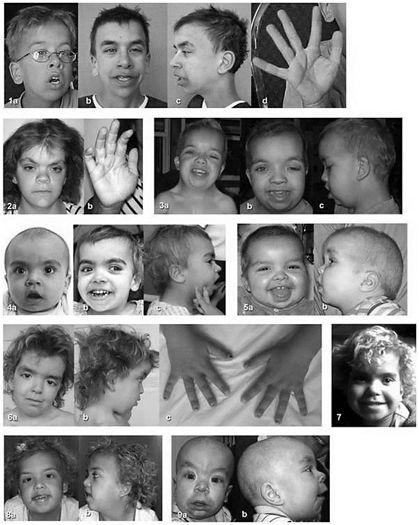
Basi Molecolari ed Eziopatogenesi
2001: IDENTIFICAZIONE DEL PRIMO GENE CAUSATIVO
Alterano l’interazione NSH2/PTP
sbilanciando l’equilibrio tra forma
inattiva e forma attiva a favore di
quest’ultima
In condizioni di riposo il dominio MUTAZIONI con guadagno di
NSH2 blocca il sito catalitico PTP funzione
- autoinibizione intramolecolare - Responsabili del 40-60% dei casi di
sindrome di Noonan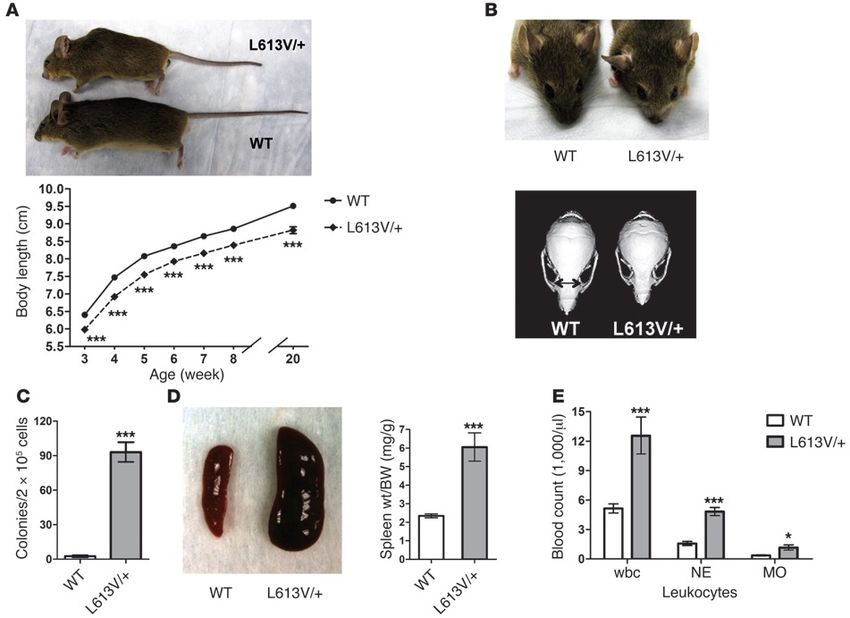
Basi Molecolari ed Eziopatogenesi
La cascata Ras/MAPK
Le mutazioni gain of
function di SHP2
determinano
iperattivazione della
cascata
?
a) Nei casi di sindrome di Noonan SENZA mutazioni di PTPN11 sono coinvolti
altri geni della pathway?
b) Condizioni cliniche simili alla sindrome di Noonan riconoscono una
patogenesi comune?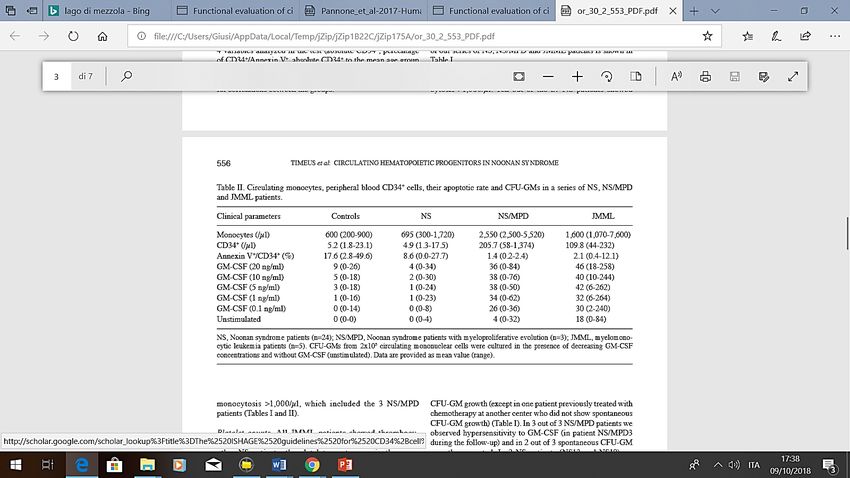
Basi Molecolari ed Eziopatogenesi a) GENI MALATTIA
2006 2010
2007
2010
2007
2015
2014
2009
2009 2016 2018
Basi Molecolari ed Eziopatogenesi
b) Sindrome di Costello, sindrome Cardiofaciocutanea, sindrome LEOPARD, sindrome di Legius, NF1:
Legius
syndrome le RASOPATIE
Sindromi da
iperattivazione
della cascata
Ras/MAPK
Il Database Rarità e grande variabilità genotipica e fenotipica à informazioni limitate e frammentarie sulla correlazione fenotipo/ genotipo e sull’evoluzione del quadro clinico Perciò è necessario raccogliere ed analizzare un maggior numero di dati a livello nazionale condividendo i set di dati clinici genetici e molecolari tra i vari Ospedali e Istituti di ricerca in Italia e nel mondo tramite uno strumento definito Database che permette attraverso il suo popolamento un vero e proprio censimento per sorvegliare la malattia. Inoltre rappresenta un importante strumento per potenziare lo sviluppo di collaborazioni scientifiche nazionali e internazionali nel settore delle malattie rare e dei farmaci. Fotografa il numero di malati per patologia e la loro distribuzione, consente di organizzare i servizi sanitari e stanziare risorse in modo preciso e mirato e, per i ricercatori, significa la possibilità di condurre studi specifici sulla malattia.
Registro Italiano della Sindrome di Noonan e delle RASopatie Obiettivo: definizione storia naturale della malattia e correlazioni genotipo-fenotipo Centri promotori Ospedale Pediatrico Bambino Gesù – Roma Ospedale Infantile Regina Margherita – Torino Fondazione Policlinico Universitario «Gemelli» – Roma Ospedale Sant’Orsola-Malpighi – Bologna Casa Sollievo della Sofferenza – San Giovanni Rotondo Università degli Studi «Federico II» – Napoli Ospedale Sant’Anna – Como Università di Padova – Padova Dati raccolti - Analisi molecolari - Storia della gravidanza - Presentazione clinica prenatale, neonatale e postnatale - Sviluppo psicomotorio - Valutazione auxologica - Immagini cliniche - Indagini biochimiche e strumentali - Valutazioni specialistiche inclusi dati nutrizionali, gastroenterologici, cardiologici, immunologici, ematologici, endocrinologici, neurologici e oncologici.
Il Database Banca Dati Nazionale, su piattaforma Cineca, per la raccolta e la gestione dei dati clinici, della storia naturale, delle complicanze, dello stato di salute a lungo termine dei pazienti affetti da sindrome di Noonan e e delle RASopatie.
Il Database – la nostra esperienza Numero
Gene pazienti
Inserimento di 136 pazienti di cui
PTPN11 80
• 80 con mutazione sul gene PTPN11
• 15 con mutazione sul gene SOS1 SOS1 13
RAF1 10
• 10 con mutazione sul gene RAF1 SHOC2 4
• 4 con mutazione sul gene SHOC2 BRAF 3
• 3 con mutazione sul gene BRAF KRAS 2
• 1 con mutazione del gene SOS2 SOS2 1
RIT1 1
• 1 con mutazione sul gene RIT1 HRAS 1
• 1 con mutazione sul gene HRAS MEK1 1
• 1 con mutazione sul gene MEK1 DIAGNOSI CLINICA 20
TOT 136
• 20 pazienti con diagnosi clinicaFenotipo postnatale
1) Cardiopatia congenita 64,4%
I = risoluzione spontanea o buon compenso senza terapia
SCORING SYSTEM Severità cardiopatia congenita II = trattamento farmacologico
III = trattamento chirurgico
IV = chirurgia non risolutiva/ trapianto cardiaco/decesso
La stenosi della polmonare più frequente nei pazienti con mutazione di PTPN11.
Non altre differenze significative nella prevalenza delle cardiopatie nei sottogruppi
mutazionaliFenotipo postnatale
2) Sviluppo neuromotorio Non differenze significative nella
distribuzione
del ritardo neuromotorio
e della bassa statura nei sottogruppi
mutazionali
0 = normale
I = ritardo delle acquisizioni neuromotorie
SCORING SYSTEM Sviluppo Neuromotorio II = difficoltà nell’apprendimento
III = difficoltà nell’apprendimento e ritardo del linguaggio
3) Accrescimento staturale
0 = normale
SCORING SYSTEM accrescimento staturale 1 = statura < 10° percentile
2 = statura < 3° percentileIdentificazione di un nuovo cluster di mutazioni di
PTPN11
Identificato un nuovo cluster di mutazioni di
PTPN11 responsabili di una forma più lieve di
Sindrome di Noonan, con minore prevalenza di
anomalie cardiache e coinvolgimento del sistema
nervoso centrale e dismorfismi facciali più
sfumati
- Identificate 5 nuove mutazioni di PTPN11 localizzate nei residui Leu261, Leu262, and Arg265 in 16 pazienti
- mutazioni che coinvolgono i residui Leu262 e Arg265 alterano l’interazione autonoibitoria N-SH2/PTP (mutazioni di
gruppo I)
- mutazioni al residuo Leu261 alterano l’attività catalitica di SHP2 (mutazione di gruppo IV)
- la mutazione p.Leu261His ha presentato effetto gain of function solo in presenza di elevate concentrazioni di EGF. In
assenza di ulteriori dimostrazioni di patogenicità, dev’essere considerata come una variante di incerto significato
(VUS)
Caso 1 Tartaglia et al. Diversity and functional consequences of
Mutazione Leu262Arg PTPN11 sporadica germline and somatic PTPN11 mutations in human
disease.Am J Hum Genet. 2006 Feb;78(2):279-90
Caso 2
Mutazione Leu261His PTPN11 familiareIdentificazione di un nuovo cluster di mutazioni di
PTPN11
CONCLUSIONI:
- eterogeneità genotipica della sindrome di Noonan
- effetto diverso delle mutazioni di PTPN11 sulla funzionalità di SHP2 e sulla
iperattivazione della cascata RAS/MAPK
- nuovo cluster identificato si associa a un fenotipo di sindrome di Noonan più sfumato
rispetto alle altre mutazioni note di PTPN11Variabilità fenotipica: il paradigma della mutazione
Ser2Gly del gene SHOC2
Studio genotipo-fenotipo in 4 pazienti con mutazione Ser2Gly di SHOC2, responsabile della «sindrome di Noonan con
Loose anagen hair»
2 casi con cardiopatia complessa, ritardo cognitivo severo, epilessia, grave stato distrofico Mutazione
2 casi con cardiopatia semplice/assenza di cardiopatia e sviluppo cognitivo adeguato p.Ser2Gly
Paziente 1. Evoluzione
fenotipica del paziente .
Quadro clinico caratterizzato
da grave stato distrofico,
cardiomiopatia ipertrofica
severa, epilessia, ritardo
cognitivo.
Paziente 2. Quadro
clinico caratterizzato da
modesto stato
distrofico. Non
cardiopatia congenita,
sviluppo
neuropsicomotorio
adeguato.Variabilità fenotipica: il paradigma della mutazione
Ser2Gly del gene SHOC2
CONCLUSIONI:
- Omogeneità genotipica può associarsi a estrema variabilità fenotipica
- Il genotipo NON predice la prognosiSignificato prognostico del fenotipo prenatale
72 pazienti 47 pazienti
• 72 pazienti con dati anamnestici prenatali disponibili
• 38/72 test di screening eseguiti: 16/38 (42%) test integrato patologico con Translucenza Nucale aumentata
• 25/72 (34,7%) polidramnios : anomalia ecografica più frequente • 6/15 (40%) idrotorace e versamenti multipli
• 5/15 (33%) anomalie cardiovascolari
• 15/72 (20,8%) anomalie ecografiche fetali patologiche • 3/15 IUGR
• 2/15 malformazioni nefro-urinarie
• 1/15 sospetta duplicazione intestinale
ANOMALIE ECOGRAFICHE FETALI • 1/15 dilatazione ventricoli cerebrali
FENOTIPO POSTNATALE p
Presenti (n = 15) Assenti (n = 57)
s 14 42
Cardiopatia congenita (CHD) 0,164
n 1 15
CHD severa (classe 3-4)
s 6 16
0,367
L’insorgenza di ritardo
n
s
9
14
41
48
neuropsicomotorio e di forme
0,674
mielodisplasiche è risultata
Bassa statura
n 1 9
s 9 34
Bassa statura severa (< 3°)
n 6 23
0,999
significativamente più frequente
s 13 26
Ritardo (classe 1-2-3)
n 2 31
0,007 nei pazienti con
JMML/MDS
s
n 9
6 0
57
< 0,001 malformazioni ecografiche fetaliSignificato prognostico del fenotipo prenatale
CONCLUSIONI:
- Polidramnios e aumento della Translucenza Nucale sono le anomalie di più frequente
riscontro in epoca prenatale, ma sono ASPECIFICHE
- Potere predittivo aumenta quando associate ad anomalie ecografiche FETALI (in
particolare cardiopatia congenita e idrotorace)
- Il riscontro di idrotrace si associa ad un aumentato rischio di sviluppare forme
mielodisplasiche e ritardo neuropsicomotorioStudio del profilo ematopoietico
- La sindrome di Noonan e la Leucemia
Mielomonocitica Giovanile (JMML) sono
caratterizzate entrambe da iperattivazione
della cascata RAS/MAPK.
- Sindrome di Noonan: mutazioni germinali di
PTPN11 in ~50% dei casi JMML (juvenile myelomonocytic leukemia) = raro disordine
- JMML: mutazioni somatiche di PTPN11 nel clonale mileodisplastico/proliferativo tipico della prima
35% dei casi infanzia caratterizzato da proliferazione spontanea in vitro di
- Differente spettromutazionale precursori ematopoietici in assenza di fattori di crescita
esogeni da ipersensibilità selettiva al GM-CSF (granulocyte-
- Analisi su linfociti di sangue periferico di 27 macrophage colony-stimulating factor)
pazienti con Sindrome di Noonan e 5 con
JMML
- Studio della conta assoluta di precursori CD
34+ , del rate apoptotico e delle CFU-GM
(crescita spontanea e dopo stimolazione con
GM-CSF) al fine di identificare sottogroppi di
pazienti Sindrome di Noonan ad aumentato
rischio di sviluppare forme mielodisplasiche
1/6 ha sviluppato una mielodisplasia nei 12 mesi
6/27 pazienti con Sindrome di Noonan hanno presentato successive, 2/6 avevano un disordine mieloproliferativo
aumentata sensibilità alla stimolazione con GM-CSF transitorio al momento dello studio
La conta assoluta di PB CD34+ nei pazienti con sindrome di Noonan è risultata normale
Rate apoptotico nei pazienti NS, NS/MDs e JMML è risultato ridotto rispetto ai controlliStudio del profilo ematopoietico
CONCLUSIONI:
- Complessità del profilo funzionale ematopoietico della sindrome di Noonan
- La Sindrome di Noonan è caratterizzata da una ridotta attività apoptotica dei precursori
ematopoietici circolanti
- Utilizzo della conta assoluta dei CD34, del rate apoptotico e delle colture cellulari CFU-
GM come metodi NON invasivi per l’identificazione precoce di soggetti ad elevato rischio
di evoluzione mieloproliferativa
Follow up ematologico miratoStudio delle caratteristiche minerali e ossee Analisi con ultrasonografia ossea quantitativa (QUS) + studio del profilo metabolico osseo in 35 pazienti affetti da Sindrome di Noonan AD-Sos (amplitude depnedent spped of sound): velocità dell’US attraverso l’osso. Correla con la densità minerale dell’osso BTT (bone trasmission time): tempo di trasmissione attraverso l’osso. Correla con densità e spessore della zona corticale. E’ indipendente dallo spessore del tessuto molle circostante. Miglior indicatore della reale condizione scheletrica. - 25% dei pazienti presentava valori di densitometria ossea ridotti rispetto alla popolazione di controllo. - Valori corretti per fattori confondenti quali età ossea, sviluppo puberale e BMI hanno confermato che nel 15% dei casi i valori di QUS si attestano al di sotto della media. CONCLUSIONI: Nella Sindrome di Noonan il coinvolgimento osseo è primitivo. L’osso è uno dei tessuti bersaglio della deregolazione della pathway.
Revisione del fenotipo cardiaco: il «CARNET study» Inclusione dei nostri pazienti nel CARNET study (CArdiac Rasopathy NETwork) : studio finalizzato a definire il rischio di morbidità e mortalità cardiaca nei pazienti affetti da Rasopatia (n= 371) Prevalenza: 298/371 pazienti con cardiopatia congenita (80.3%). La Stenosi Polmonare è la più comune delle cardiopatie congenite (59%), seguita dalla Cardiomiopatia Ipertrofica (27%) e dai difetti settali atriali (11%) Morbidità: 141/298 pazienti (47.3%) è stata sottoposta ad almeno un intervento cardiochirurgico o percutaneo, soprattutto pazienti con Stenosi Polmonare e mutazione di PTPN11. Mortalità: Mortalità bassa nei pazienti affetti da Rasopatia. 10/371 decessi (2.7% dell’intera coorte, 3.4% dei pazienti con coinvolgimento cardiaco), di cui 8/10 per cause cardiache. Aumentata incidenza di complicanze intraoperatorie letali e morte improvvisa nei pazienti PTPN11 con Cardiomiopatia Ipertrofica.
Prospettive future – Analisi esomica nei pazienti con analisi molecolare standard e array-CGH negativo per la ricerca di nuovi geni malattia (15% dei pazienti con diagnosi clinica) – Studiare eventuali concause genomiche della variabilità fenotipica mediante array- CGH nei pazienti con diagnosi molecolare positiva e fenotipo particolarmente severo, nell’ipotesi che la deregolazione della cascata RAS/MAPK sia condizione necessaria ma non sufficiente per il manifestarsi di determinate caratteristiche nei soggetti affetti – Stratificazione delle caratteristiche cliniche auxologiche (parametri neonatali, crescita e pubertà) sulla base dei geni coinvolti – Valutazione NPI approfondita di tutti i pazienti afferenti alla Clinica Pediatrica dell’Ospedale Infantile Regina Margherita di Torino – Approccio terapeutico con inibitori della cascata RAS/MAPK (MEK inibitori)
Mek inibitori - La nostra esperienza…
29/12 35+2 settimane 3670 g
Riscontro prenatale di polidramnios e cardiomiopatia ipertrofica
Alla nascita evidenti caratteristiche faciali, desaturazioni e necessità di
intubazione endotracheale e ventilazione meccanica
Ecocardioà evidente cardiomiopatia ipertrofica
02/01 Fase di ipertensione polmonare con ventilazione in alta frequenza,
05/01 Evidenziata emorragia ventricolare ostruttiva à drenaggio
12/01 miglioramento della dinamica respiratoria, estubazione
Avvio degli accertamenti genetici per sospetta RASopatiaMek inibitori - La nostra esperienza…
12/01 stabilità del quadro clinico (respiro spontaneo in aria ambiente) e
progressione della cardiomiopatia ipertrofica
29/01 Successivo progressivo scompenso cardiaco, prescrizione di B-bloccanti e
diuretici, restrizione idrica
09/02 A 1 mese e mezzo scompenso grave, reintubata, aggiunta di inotropi,
trasferimento in rianimazione, intubazione e ventilazione in alta
frequenza
11/02 Terapia massimale per scompenso cardiaco non controllatoMek inibitori - La nostra esperienza… Conferma molecolare di RASopatia, mutazione RAF1 p.Ser257Leu (770C > T, exon 7) Mutazioni in RAF1 sono associate specificamente a cardiomiopatia ipertrofica Mutazione RAF1 p.Ser257Leu associata a cardiomiopatia ipertrofica grave e ipertensione polmonare
Sindromi, gene mutato e cardiopatia tipica
~7%
~70%
~70%
~85% ~35%
~50%Cardiomiopatia ipertrofica
VS
VD
Cuore normale Cardiomiopatia ipertrofica
Normale Apicale Settale (ostruttiva) Globale (ostruttiva)Sopravvivenza, cardiomiopatia ipertrofica e
scompenso cardiaco
Survival by age and congestive heart
failure in children with Noonan syndrome
and hypertrophic cardiomyopathy
I neonatiIperattivazione RAS-MAPK pathway Negli ultimi 10 anni, mutazioni germinali di numerosi geni della via RAS- MAPK sono stati identificati come responsibili delle RASopatie La RAS- MAPK pathway è una cascata di segnale centrale in biologia unana, può essere attivata da una serie di agonisti ed è presente in tutti i sistemi cellulari, incluse le cellule cardiache (miociti)
Modello murino con mutazione RAF1 L613V Effetti benefici a livello cardiaco ed extracardiaco in modelli preclinici murini
Modello murino con mutazione RAF1 L613V
Bassa statura Caratteristiche
facciali Cardiomiopatia ipertrofica
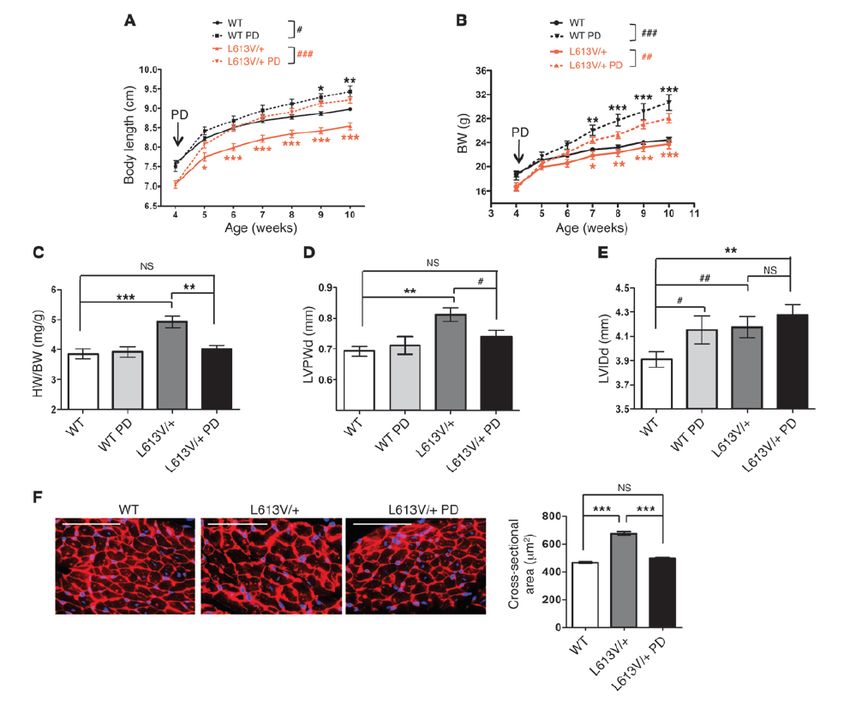
MielodisplasiaTrattamento con MEK-inibitore
- Aumento di peso e lunghezza
- Normalizzazione del loro rapporto
- Normalizzazione spessore parete
cardiaca, dimensioni, anatomia e
funzionalità
- Riduzione delle caratteristiche
facialiMEK inibitori TRAMETINIB inibitore allosterico reversibile altamente selettivo di MEK1/2, è approvato dal 2013 dall’FDA per il trattamento di tumori specifici con attivazione della via RAS/MAPK e presenta rapido effetto e lunga emivita
RASopatie e Melanoma
La cascata RAS/MAPK è una
via di segnalazione
intracellulare coinvolta nel
regolazione della
proliferazione cellulare e
sopravvivenza del delle
cellule tumorali.
Nei melanomi e in altri
tumori solidi, diverse
mutazioni di BRAF o NRAS,
esercitato un effetto
oncogenico attivando la
cascata, con conseguente
aumento della
proliferazione cellulare. Mutazioni in geni della RAS-MAPK pathway
condivise tra RASopatie e MelanomaRASopatie e Melanoma
Queste mutazioni sono diventate bersagli di nuove strategie terapeutiche nel
melanoma e altri tumori.
Inibitori i BRAF e
di MEK hanno la
capacità di
inibire la crescita
e indurre la
cellula morte
×
nelle linee
cellulari di
×
melanoma.
Attualmente in uso una strategia terapeutica
con inibitore di BRAF (DABRAFENIB) e di MEK
(TRAMETINIB)Utilizzo clinico MEK inibitori
2 pazienti con S. di Noonan
RIT1 c.104G>C; p.Ser35Thr
RIT1 c.246T>G, p.Phe82Leu
Cardiomiopatia progressiva nonostante B-blocco
Ventilazione meccanica dalla nascita
• Riduzione ipertrofia cardiaca e dell’ostruzione valvolare in 4 mesi
• Rapida risposta clinica (miglioramento edema e estubazione)
• Risposta laboratoristica (normalizzazione di NT-proBNP)
• Crescita corporea
(da miglioramento
dello scompenso
cardiaco?)
• In un paziente
regressione del
versamento
chiloso pleuricoMek inibitori - La nostra esperienza… Vista la criticità del quadro clinico di scompenso cardiaco congestizio terminale viene proposta ai genitori terapia con Trametinib off-label Inizia Trametinib 0,025 mg/kg/die per os. Protocollo di monitoraggio • NT-proBNP ogni 2 giorni • Ecocardio ogni 3 giorni • ECG settimanale (prolungamento QT) • Peso quotidiano, Lunghezza + Circonferenza cranica settimanale • PAO (segnalata ipertensione) • Emocromo settimanale • Funzione epatica (AST ALT GGT LDH CK PT PTT INR F dD) 15 giorni • Dosaggio farmaco settimanale +2 giorni: riduzione edema, miglioramento dinamica respiratoria +6 giorni: estubazione +6-20 giorni stabilità con progressiva riduzione supporto respiratorio fino a solo O2 …
Istologicamente quadro di grave ipertensione polmonare
Conclusioni
• I dati preliminari sull’utilizzo di farmaci inibitori di MEK sono
incoraggianti
• La nostra esperienza di difficile interpretazione:
• Buona risposta iniziale
• Presenza di fattori aggravanti (emorragia cerebrale e
aneurisma della polmonare)
• Indispensabili studi più approfonditi
- Diversa risposta in diverse mutazioni?
- Età per iniziare la terapia?
- Mancanza dati su terapia a lungo termine
- Dose ottimale?
- Quanto tempo proseguire la terapia?
- Effetti collaterali?Ringraziamenti
Prof. Giovanni Battista Ferrero
Dott.ssa Giuseppina Baldassarre
Dott. Alessandro Mussa
Prof. Marco Tartaglia
Prof. Gregor AndelfingerPuoi anche leggere

















































