Università degli Studi di Napoli "Federico II"
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Università degli Studi di Napoli “Federico II”
Scuola Politecnica e delle Scienze di Base
Area Didattica di Scienze Matematiche Fisiche e Naturali
Dipartimento di Fisica “Ettore Pancini”
Laurea triennale in Fisica
La citometria di flusso come tecnica avanzata di analisi
in biofisica
Relatori: Candidato:
Prof. Lorenzo Manti Alessandra Ippolito
Dott. Valerio Ricciardi Matricola N85001042
A.A. 2020/2021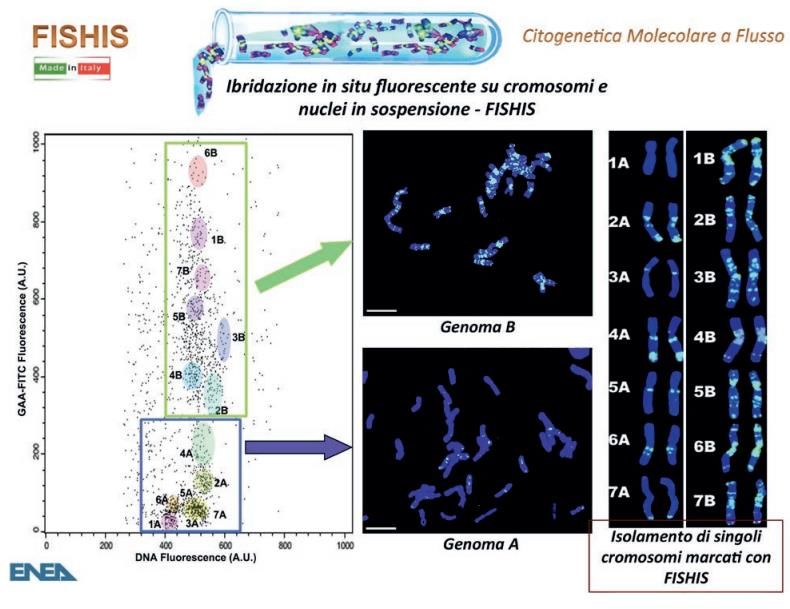
INDICE
Introduzione……………………………………………………………3
1. La citometria a flusso nella Biofisica…………………………5
1.1. Cenni storici e applicazioni in Biofisica…………………………………5
1.2. Tecniche citofluorimetriche…………………………………………………8
1.3. Strumentazione…………………………………………………………………..10
1.4. Costruzione di pannelli di citometria a flusso……………………..13
2. Proliferazione cellulare mediante citometria a
flusso……………………………………………………………………...17
2.1. Danno da radiazione al DNA……………………………………………...17
2.2. Modifiche apportate dalla radiazione sul ciclo cellulare…....19
2.3. Misurazione della sintesi del DNA………………………………………21
2.4. Monitoraggio di generazioni di divisione cellulare……………..24
3.Saggi di apoptosi per citometria a flusso………………….26
3.1. Danno da radiazione e apoptosi…………………………………………27
3.2. Colorazione dell’annessina V……………………………………………..29
3.3. Saggi di apoptosi dell'attività della caspasi per la citometria
a flusso.……………………………………………………………………………..31
3.4. Saggi di funzione dei mitocondri………………………………………..33
3.5. Saggi di apoptosi nucleare per citometria a flusso……………..34
Conclusioni…………………………………………………………………37
Bibliografia…………………………………………………………………38
Ringraziamenti……………………………………………………………41
2
Introduzione
La citofluorimetria è una tecnica di laboratorio che consente di rilevare, identificare e
contare specifiche cellule. Questo metodo è anche in grado di identificare dei
componenti particolari contenuti all'interno delle cellule.
La citometria si divide in due aree, le quali si distinguono in base alla preparazione dei
campioni: citometria statica, nella quale l’elemento cellulare viene prima visualizzato
dall’operatore e poi analizzato e citometria a flusso (CFM), nella quale elementi
cellulari vengono iniettati in un filetto liquido, il quale tende, in condizioni
idrodinamiche opportune a trasportare le cellule in maniera separata ed ordinata in
un punto in cui arriva un fascio di luce focalizzata.
Per quanto riguarda sia la citometria a flusso che la statica, la finalità è quella di
misurare parametri biofisici e biochimici per poi raccoglierli nella memoria di un
microprocessore, rappresentarli graficamente mediante opportuni software e
analizzarli con metodi statistici [1].
Le informazioni riguardo i campioni cellulari analizzati, possono essere raccolte grazie
a dei protocolli di identificazione che prevedono la valutazione delle caratteristiche
fisiche delle cellule ma anche della presenza o assenza di particolari marcatori,
gli antigeni, presenti sulla superficie o all'interno delle cellule stesse.
Lo spettro delle grandezze o caratteristiche cellulari, che oggi è possibile misurare
mediante citometria a flusso, è assai ampio e va dalla determinazione dei parametri
quali volume, massa di sostanze (DNA, RNA, proteine, per citare le principali), alla
funzione (espressione di antigeni di membrana ed endoplasmatici, determinazione
della concentrazione degli ioni calcio, misura del pH, ecc.).
Si può quindi dedurre che tale tecnica ha apportato innumerevoli vantaggi per quanto
riguarda lo studio cellulare. L’analisi multi-parametrica effettuata mediante i moderni
citometri a flusso, permette infatti la misurazione simultanea di molteplici
caratteristiche cellulari, senza dover ricorrere a strumentazioni più complesse e
laboriose.
Durante la trattazione di tale tecnologia, ci soffermeremo maggiormente sulle
applicazioni di quest’ultima in ambito biofisico, specificando alcuni particolari
endpoint di interesse per gli effetti dell’azione della radiazione a livello cellulare.
La tesi si divide in tre capitoli: nel primo è descritta la citometria, con accenni storici
sulla sua nascita ed evoluzione, le varie tecniche esistenti e la strumentazione
utilizzata.
Il secondo capitolo riguarda, invece, l’applicazione della citometria allo studio della
proliferazione cellulare.
Nell’ultimo capitolo sono invece descritti i saggi, effettuati tramite citometria a flusso,
in grado di rilevare i processi di apoptosi cellulare, uno dei possibili meccanismi di
3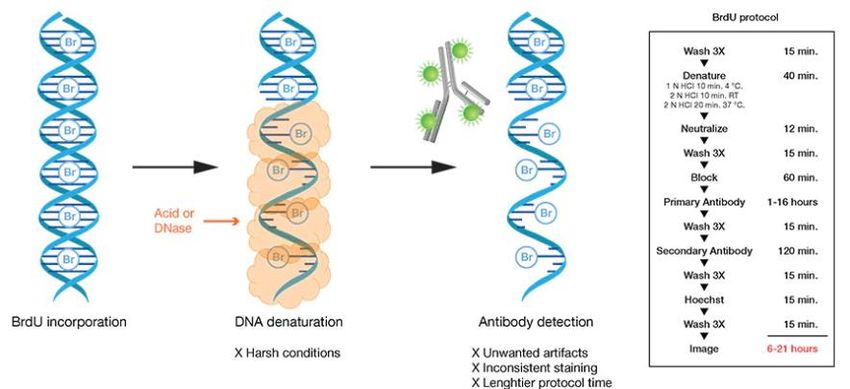
morte cellulare a seguito dell’esposizione a radiazioni, particolarmente rilevante in
ambito biofisico.
4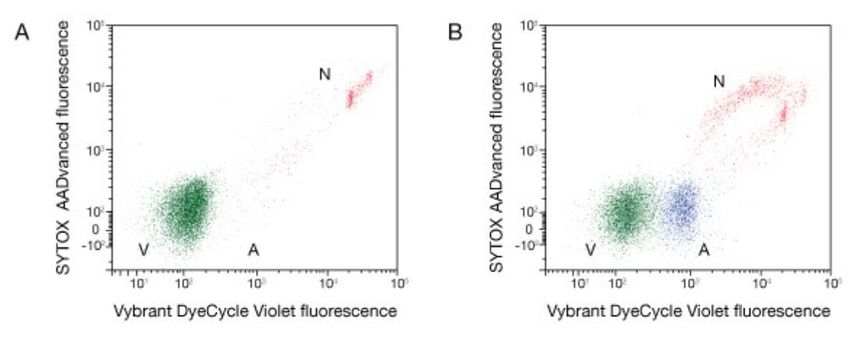
1 La citometria a flusso nella Biofisica
1.1 Cenni storici e applicazioni in Biofisica
Per citometria a flusso si intende una tecnica mediante la quale analizzare una
popolazione di cellule in sospensione liquida tramite un opportuno strumento
denominato citometro a flusso.
La comparsa della citometria a flusso (CFM) avviene intorno alla fine degli anni
Sessanta e determina un veloce ed intenso sviluppo delle tecniche istologiche e
citochimiche, dando così un impulso decisivo soprattutto agli studi sulla proliferazione
cellulare.
Precursore di tale tecnica fu lo scienziato M.J. Fulwyler, il quale sviluppò un dispositivo
in grado di separare le cellule biologiche (sospese in un mezzo conduttore) in base al
volume [2].
In questa prima versione di citometro, del 1965, il volume cellulare era misurato in
un'apertura di Coulter, ovvero un tubo di vetro cavo con un foro attraverso il quale le
cellule venivano trascinate nel contaglobuli (Figura 1). Quest’ultimo, era riempito di
una soluzione salina ed al suo interno era generata una corrente elettrica grazie alla
presenza di due elettrodi, uno interno e uno esterno al tubo stesso. Durante la misura
di un campione, il tubo era posto sottovuoto e le cellule transitavano, attraverso il
foro, all’interno di esso. Ogni volta che una cellula (o particella) entrava nel foro
dell'apertura, essa spostava la soluzione salina generando in un impulso elettrico
visibile. In base all’intensità di quest’ultimo era possibile trarre informazioni riguardo
le dimensioni della cellula.
Figura 1. Esempio di Aperture tube [3].
Successivamente, le cellule erano isolate in goccioline del mezzo caricate
elettricamente in base al volume rilevato. Le goccioline cariche erano poi deviate da
un campo elettrostatico in un recipiente di raccolta.
Utilizzando questo apparato miscele di eritrociti di topo ed umani, e cellule di linfoma
di topo sono state separate con successo; nei test con cellule ovariche di criceto cinese
5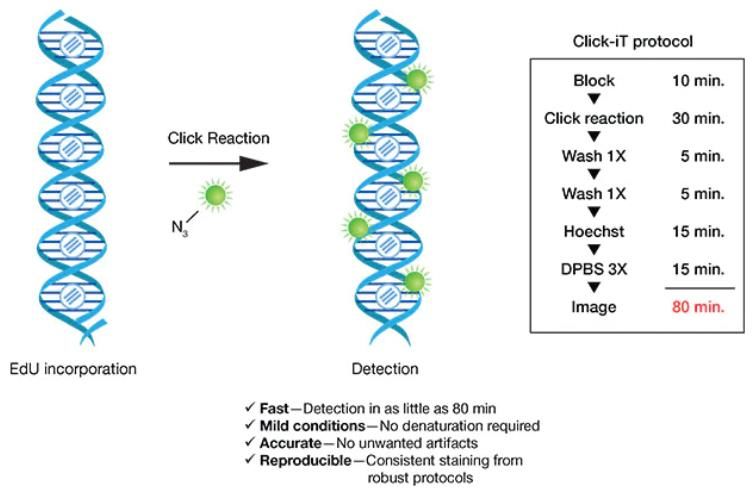
essenzialmente tutte le cellule sono sopravvissute alla separazione e sono cresciute
al loro ritmo normale.
Risale invece al 1968 Il primo dispositivo di citometria a flusso basato sulla
fluorescenza [4].
Al giorno d’oggi, in generale, possiamo dire che la citometria a flusso è la tecnica
mediante la quale diverse cellule passano singolarmente attraverso una camera di
flusso e vengono esposte alla luce o alla corrente elettrica. In base alle dimensioni ed
alla granularità intracellulare o alla strutturazione delle cellule, si registreranno gli
effetti sulla trasmissione elettrica, sull’assorbimento della luce, sulla fluorescenza e
sulla riflessione.
L’elaborazione di queste informazioni da parte di un computer, che analizza un
numero elevato di cellule, permetterà poi di misurare diversi parametri di
fluorescenza in base ai dati raccolti dai tubi fotomoltiplicatori.
Tra i possibili parametri misurabili abbiamo: il contenuto proteico e di DNA, ovvero
possibili variazioni del contenuto di DNA nelle cellule figlie in seguito al processo di
sintesi; contenuto di RNA, ovvero possibili mutazioni del contenuto di RNA durante la
fase G1 del ciclo cellulare; l’immunofenotipo, ovvero la caratterizzazione degli antigeni
espressi sulla membrana cellulare; gli organelli intracellulari, ovvero il riuscire a
valutare l’attività mitocondriale; attività enzimatica, ovvero lo studio della velocità
con cui una reazione procede verso l’equilibrio in presenza di un enzima.
Tutto ciò è possibile mediante l’utilizzo di fluorocromi, i quali, essendo sostanze che,
quando eccitate da una sorgente luminosa, emettono segnali fluorescenti, possono
individuare specificamente antigeni, marcatori e proprietà biochimiche cellulari.
I più moderni fotomoltiplicatori sono capaci, inoltre, di ordinare le cellule in base alle
proprietà desiderate basandosi sulla deflessione delle gocce che queste cellule
contengono.
Fin dall’inizio la CFM si prefigge lo scopo di misurare proprietà multiple di singole
cellule ad una velocità così rapida da permettere una dettagliata analisi qualitativa e
quantitativa in tempi molto brevi.
Inizialmente la CFM era limitata alla misura di uno o due parametri: generalmente, un
parametro per la misura fisica del light-scattering e l’altro per fluorescenza. La
strumentazione era caratterizzata da varie sorgenti di eccitazione. Tipicamente gli
immunologi si avvalsero di antisieri e anticorpi monoclonali (MoAb) marcati con
isotiocianato di fluoresceina (FITC), un composto organico fluorescente in grado di
legarsi ai gruppi amminici e solfidrilici delle proteine permettendone la visualizzazione
in fluorimetria.
Risultò presto evidente che molti MoAb avevano reazioni sovrapposte con vari
subsets cellulari. Questo, aggiunto alla complessità del sistema immunitario, rivelata
dall’uso di questi MoAb diretti contro antigeni (Ag) di superficie linfocitaria, stimolò
sia lo sviluppo di MoAb sempre più specifici, sia la ricerca di nuovi reagenti
6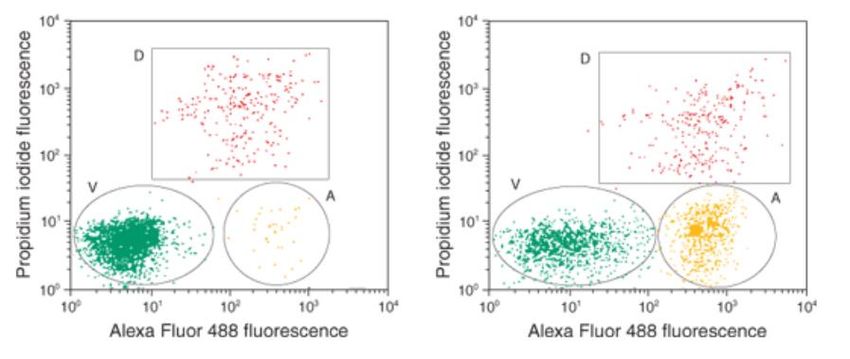
fluorescenti e di tecniche per coniugarli a tali Ab, nonché di citometri a flusso multi-
parametrici più sensibili.
Il problema principale fu quello di trovare reagenti che potessero essere coniugati agli
Ab senza che venisse modificata la loro capacità di legame e di selezionare
fluorocromi con spettri distinti di emissione. I primi sistemi in CFM a doppia
fluorescenza impiegarono Ab coniugati con fluoresceina e rodamina, ma ne risultava
una sovrapposizione spettrale considerevole (Figura 2). Il problema fu superato con
la sintesi di derivati della rodamina, quali il Texas-red (C31H29ClN2O6S2) [5], colorante
fluorescente impiegato con successo in combinazione con il FITC, anche se erano
necessarie due sorgenti di eccitazione, rispettivamente a lunghezze d’onda pari a 600
e 488 nm.
Figura 2. Grafici degli spettri di eccitazione ed emissione in fluorescenza della fluoresceina (A) e della
rodamina (B), rispettivamente. La curva che rappresenta l'emissione (in rosa), nello spettro dei fluorocromi, è
usualmente più bassa o uguale in intensità rispetto alla curva di eccitazione (in blu) [6].
Un punto di svolta nella CFM fu lo sviluppo di coloranti quali le ficobiliproteine [1].
Questi fluorocromi naturali sono solubili in acqua, caratterizzati da un pH neutro,
facilmente coniugabili con MoAb ed hanno rese quantiche molto elevate.
7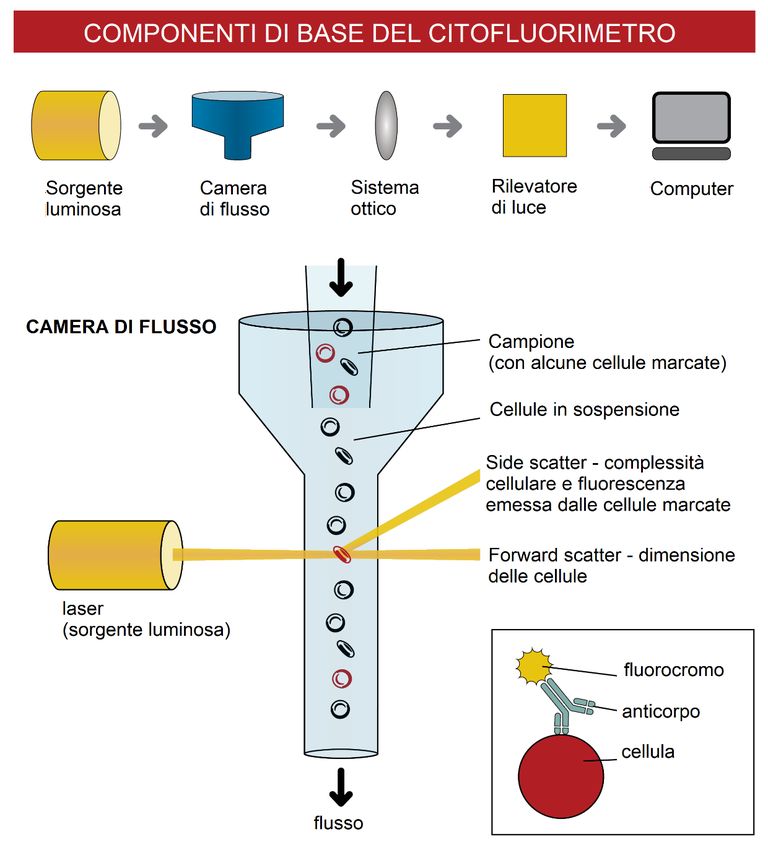
Ruolo fondamentale, per superare i limiti del passato, risiede oggi nella disponibilità
di MoAb marcati con fluorocromi e diretti contro una larghissima varietà di Ag di
membrana e/o intracellulari, che identificano la linea cellulare, la composizione in
sottopopolazioni, il livello di differenziazione e le caratteristiche associate alla
proliferazione.
Sino a qualche anno fa, con l’analisi biochimica classica, utilizzando estratti cellulari,
si perdevano un gran numero di informazioni riguardo l’assegnazione di uno specifico
parametro ad una cellula o, in generale, sulla distribuzione dei parametri. Al giorno
d’oggi, invece, grazie ai progressi tecnologici per questa tecnica, si hanno informazioni
sempre più specifiche e chiare riguardo i parametri cellulari.
Lo studio della fisiologia e del comportamento delle cellule ha trovato quindi in questa
tecnica un sostegno insostituibile. Infatti, in pochi secondi è possibile analizzare da
10.000 a 20.000 cellule quantificando numerosi parametri per ogni singola cellula.
Tra i parametri che è possibile analizzare troviamo il contenuto proteico e di DNA, il
contenuto di RNA, l’immunofluorescenza (utilizzando particolari anticorpi è possibile
caratterizzare ad esempio i sottotipi di linfociti), gli organelli intracellulari e l’attività
enzimatica quale quella della b-galattosidasi, della glucosidasi, della glucuronidasi,
delle esterasi e delle fosfatasi.
1.2 Tecniche citofluorimetriche
Principio fondamentale alla base della citometria a flusso è la dispersione della luce.
Il citometro, tramite un flusso laminare, organizza le cellule in un flusso ordinato così
da portare queste ultime nella camera di conta, all’interno della quale avverrà l’analisi
di ogni singola cellula.
Le tecniche citofluorimetriche prevedono dunque diverse fasi.
Come step iniziale, viene preso un campione di cellule, il quale viene trattato con
reagenti specifici, principalmente fluorocromi fluorescenti legati ad anticorpi
monoclonali diretti verso particolari distretti cellulari o antigeni marcatori, che
permettono di discriminare i sottotipi cellulari.
Il campione viene poi sospeso all’interno di un fluido ed introdotto nello strumento
chiamato citofluorimetro. Qui il fluido contenente le cellule viene incanalato nella
camera di flusso.
La camera di flusso contiene uno o più laser e più rilevatori in grado di identificare
alcune caratteristiche, uniche per ciascuna cellula. Ciascun laser incide sulle cellule
presenti nel flusso, generando per ciascuna di esse uno fenomeno di scatter, la cui
intensità è dipendente dalle caratteristiche della stessa, che può essere acquisito da
uno o più rilevatori posti nelle sue vicinanze (Figura 3); il sistema così composto è in
8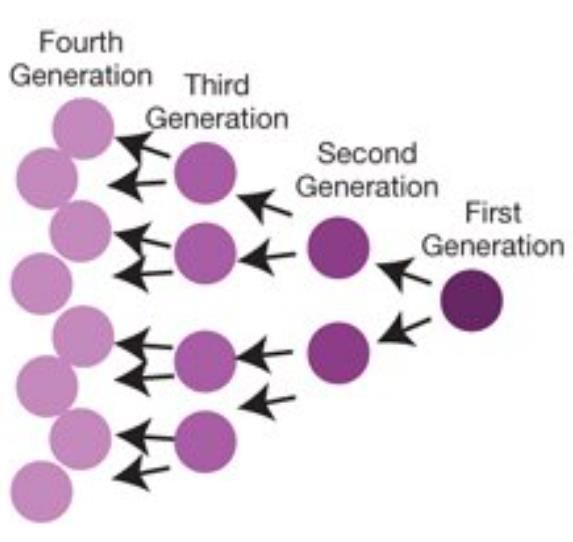
grado di analizzare ciascuna cellula presente all'interno del flusso ad una velocità
altissima (da centinaia a migliaia di cellule per secondo).
Figura 3. Schema delle componenti e del meccanismo di funzionamento di un citometro a flusso.
Le caratteristiche rivelate possono essere fisiche (dimensione e complessità cellulare)
o possono dipendere dal segnale generato dai differenti fluorocromi a seguito
dell’eccitazione causata dalla luce laser. La combinazione di queste informazioni
genera un profilo caratteristico per ciascuna cellula presente all'interno del campione.
Il segnale rilevato dai rilevatori viene amplificato dai fotomoltiplicatori e inviato al
computer. Qui viene convertito in formato digitale e mostrato sul computer o
stampato.
I dati sono, in genere, mostrati sotto forma di grafici.
9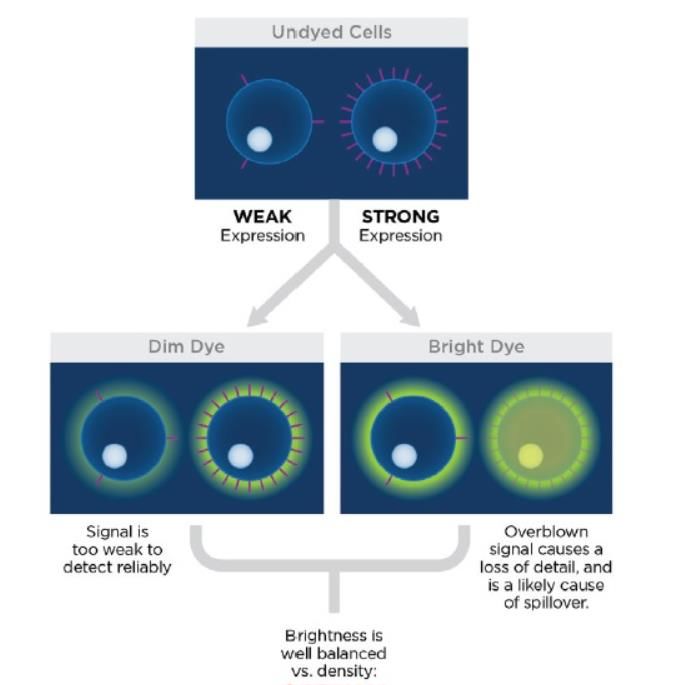
Questa analisi permette, dunque, la valutazione delle caratteristiche e del numero
delle cellule presenti nel campione.
Tra le tecniche più avanguardistiche utilizzabili con un citometro a flusso troviamo
l’ibridazione fluorescente in situ in sospensione (Fluorescence In Situ Hybridization In
Suspension, FISHIS). Quest’ultima consente di marcare il DNA di nuclei e cromosomi
in sospensione con sonde molecolari a DNA, coniugate con coloranti fluorescenti, per
identificare immediatamente e con precisione alterazioni del patrimonio genetico,
riconosciute tramite citometria a flusso (Figura 4).
Figura 4. Esempio di risultati ottenibili utilizzando la tecnica FISHIS [7].
1.3 Strumentazione
Un citometro a flusso presenta diverse componenti, ognuna delle quali svolge
un’azione ben precisa:
Il sistema fluidico: Il sistema di dispersione del campione liquido fornisce un
efficiente mezzo in grado di presentare individualmente le cellule del campione
alla stazione di misura, dove intersecano il raggio di luce emesso dal laser.
Camera a flusso: Le cellule mono-disperse vengono aspirate in una camera a
flusso, dove vengono diluite e allineate tramite un sistema fluidico a flusso
laminare.
10 Sorgenti luminose: Nella grande maggioranza dei citofluorimetri si utilizza
come sorgente luminosa un laser a ioni di Argon centrato su una lunghezza
d’onda di 488 nm (blu). Tale luce consente una efficace misura dei parametri
fisici, inoltre permette la contemporanea eccitazione di diversi fluorocromi;
sono disponibili anche citometri equipaggiati con due o più laser a diverse
lunghezze d’onda.
Parametri fisici: quando viene colpita dal fascio di luce emesso dal laser, la
cellula emette segnali di luce diffusa in base alle proprie caratteristiche fisiche
e morfologiche, per fenomeni di rifrazione, riflessione, e diffrazione. In
particolare, la luce dispersa in avanti (forward scattering, FSC) è legata alle
dimensioni delle cellule, mentre la luce riflessa a 90° (side scattering, SSC) è da
attribuire a parametri della morfologia cellulare come la granulosità del
citoplasma, il rapporto nucleo/citoplasma o la rugosità di superficie.
Citogramma: diagramma bidimensionale che permette di discriminare tra
diverse popolazioni cellulari basandosi solamente sulle loro caratteristiche
fisiche.
Segnali di fluorescenza: in citofluorimetria vengono rilevati segnali fluorescenti
generati da fluorocromi che si legano a determinate sostanze come DNA e RNA.
I dati sulla fluorescenza vengono raccolti generalmente allo stesso modo dei
dati a dispersione laterale. In una popolazione di cellule marcate, alcune
saranno più luminose di altre. Quando ogni cellula attraversa il percorso del
laser, viene generato un segnale di fluorescenza. La luce fluorescente viene
quindi diretta al rivelatore appropriato dove viene tradotta in un impulso di
tensione proporzionale alla quantità di fluorescenza emessa. Tutti gli impulsi di
tensione vengono registrati e possono essere presentati graficamente. Ogni
fluorocromo presenta una caratteristica lunghezza d’onda per l’eccitazione e
l’emissione. È possibile utilizzare più fluorocromi con emissioni differenti su un
citometro a flusso e il numero di segnali rilevabili dipende dal numero di
rivelatori disponibili nel citometro. I diversi colori vengono raccolti utilizzando
filtri ottici selezionati che dirigono la luce al rilevatore giusto e catturano i
segnali fluorescenti di picco.
Tra i vari dispositivi adoperati per realizzare tale tecnica sopracitata, ci soffermeremo
maggiormente sullo strumento disponibile presso il Laboratorio di Biofisica delle
Radiazione del Dipartimento di Fisica dell’Università “Federico II” di Napoli, ovvero il
citometro a flusso Attune™ NxT blue (ThermoFisher Scientific, Waltham, MA, USA).
Tale dispositivo presenta una frequenza di 50/60 Hz e laser di eccitazione nel blu della
lunghezza d’onda di 488 nm, con una potenza di 50 mW.
11Caratteristica particolare di tale dispositivo sta nella combinazione di tecnologia di
messa a fuoco acustica abbinata alla tradizionale messa a fuoco idrodinamica, che
permette una velocità di acquisizione fino a 10 volte più veloce rispetto a citometri a
flusso con messa a fuoco tradizionale; permette di contare, infatti, 35.000 eventi al
secondo e ha una velocità elettronica massima di 65.000 eventi al secondo.
Altra peculiarità del citometro a flusso Attune NxT è l’utilizzo di un laser flat-top,
ovvero un laser con un profilo di intensità che è piatto sulla maggior parte dell'area
coperta e che consente una finestra molto più ampia di allineamento (Figura 5).
Figura 5. Profili di emissione dei laser utilizzati nei citometri a flusso: (A) Profilo laser gaussiano con
allineamento corretto, (B) Laser gaussiano profilo con disallineamento, (C) profilo laser flat-top con
allineamento corretto, e (D) profilo laser a sommità piatta ancora allineato correttamente [8].
Tale strumento può essere utilizzato per tutti i principali studi di
immunofenotipizzazione e segnalazione, analisi del ciclo cellulare, rilevamento di
eventi rari, analisi delle cellule staminali, studi sul cancro e sull'apoptosi, saggi
microbiologici, ecc.
Per quanto riguarda i rivelatori alcuni fotomoltiplicatori sono dedicati alla
fluorescenza e al rilevamento SSC e l'FSC è ottenuto tramite rivelatore a diodi. Il
dispositivo (Figura 6), dispone di 5 diversi rivelatori per i segnali fluorescenti e di SSC,
centrati rispettivamente sulle lunghezze d’onda 488, 530, 574, 695 e 780 nm che i
segnali possono raggiungere tramite un apposito sistema di filtri (Figura 7).
12Figura 6. Citometro a flusso Attune™ NxT blue presento per il Laboratorio di Biofisica delle Radiazioni presso
il Dipartimento di Fisica dell’Università “Federico II” di Napoli [8].
Figura 7. Schema dei diversi rivelatori e filtri presenti sul citometro a flusso Attune™ NxT blue; per i diversi
rivelatori, numerati da 1 a 5 sono riportate lunghezza d’onda centrale ed ampiezza dell’intervallo di
rivelazione; un sistema di filtri passa alto (long-pass, LP) e dicroici passa alto (dichroic long-pass, DLP)
permette il passaggio del segnale verso i diversi rivelatori [8].
1.4 Costruzione di pannelli di citometria a flusso
Tramite l’utilizzo di pannelli, ovvero un insieme di fluorocromi e reagenti, ben
selezionati, la citometria a flusso multicolore è un potente strumento per rilevare e
monitorare più caratteri del campione contemporaneamente.
Ovviamente la qualità dei dati dipende, maggiormente, dall'ottimizzazione del design
del pannello e dalla corretta regolazione della strumentazione.
Per realizzare un pannello consono al trattamento di tali dati, è necessario tener conto
di diversi aspetti.
Come prima cosa bisogna selezionare i giusti fluorocromi. Essi, infatti, non sono tutti
uguali; alcuni possono essere più fluorescenti, altri più tenui. In generale, la scelta
13migliore consiste nell’utilizzare un marcatore sufficientemente brillante da rilevare
l'antigene di interesse, causando al contempo il minimo spillover o scarico in altri
rivelatori.
Con il termine spillover si descrive il fenomeno per cui lo spettro di emissione di un
fluorocromo viene rilevato da un rivelatore impostato per misurare l'emissione di un
diverso fluorocromo in maniera indesiderata, contribuendo così a un segnale
indesiderato nel canale del secondo fluorocromo. Un'emissione più brillante significa
un maggiore potenziale di spillover e una maggiore diffusione dei dati, che può
portare a una perdita di risoluzione. Per evitare ciò, si sceglie di utilizzare coloranti più
brillanti per gli antigeni scarsamente espressi, mentre, per gli antigeni fortemente
espressi, coloranti più tenui (Figura 8).
Figura 8. Differenza di emissione di fluorocromi. A sinistra è mostrato un antigene scarsamente espresso
associato ad un colorante più brillante. A destra si accosta ad un antigene fortemente espresso un colorante
più tenue.
Altro aspetto importante è l’esclusione dell’antigene. Infatti, lo spillover tra gli spettri
di emissione degli antigeni non interferirà con il corretto funzionamento del pannello
se gli antigeni non sono espressi insieme sulle stesse cellule nella porta cellulare di
interesse (Figura 9).
Affinchè tale fenomeno di spillover sia minimo, si utilizza il processo di
“compensazione”. Quest’ultimo rimuove il segnale di un dato fluorocromo da tutti i
rivelatori ad eccezione di quello impiegato per misurare quel determinato colorante.
La compensazione è il processo mediante il quale lo "spillover" di fluorescenza tra i
rivelatori viene corretto matematicamente.
14Il sistema a due rivelatori, ad esempio, può essere progettato per discriminare la
fluorescenza FITC dalla fluorescenza PE (ficoeritrina).
Quello che accade è che la luce di emissione viene suddivisa in base alla lunghezza
d’onda e, di conseguenza, distribuita ai rivelatori, ognuno dei quali ha un diverso filtro
che elimina la luce all’interno di tutto lo spettro tranne che in una regione definita di
quest’ultimo.
Ad esempio, la fluorescenza del FITC è predominante in un rivelatore con filtro da 530
nm, mentre quella della PE in un rivelatore con filtro da 575 nm; tuttavia, nel
rivelatore PE appare una certa fluorescenza FITC a causa della sovrapposizione di
emissione di questi due fluorocromi.
Per quanto detto, ogni volta che sarà presente segnale dovuto a FITC, questo
contribuirà ad un segnale nella banda dei 530 nm, ma anche ad uno nella banda dei
575 nm. PE, invece, contribuirà esclusivamente ad un segnale nella banda dei 575 nm.
Per misurare quanto vale l’intensità del segnale di FITC e quanto quello di PE nella
banda dei 575 nm, si usa il principio della compensazione: ovvero, si preparano diversi
campioni del materiale da misurare, marcati con i singoli florocromi e si misura la
quantità di segnale indesiderata presente nei diversi rivelatori; tale quantità potrà poi
essere sottratta tramite software nelle misure effettuate con presenza in
contemporanea di entrambi i coloranti.
Questo tipo di compensazione è detto “unidirezionale”: la correzione per l’emissione
di un fluorocromo in un secondo rivelatore [9].
Figura 9. Esempio di spillover non interferente con il corretto funzionamento del pannello. Gli antigeni non
vengono espressi simultaneamente sulle stesse cellule.
Un altro aspetto importante di cui tenere conto è la co-espressione antigenica.
Se un marcatore ha una debole densità di espressione lo spillover potrebbe occludere
i dati e mascherare le potenziali modulazioni. Per evitare ciò, si deve ridurre lo
15spillover dalle etichette dei fluorocromi degli antigeni co-espressi nel canale di tale
marcatore.
Solo lo spillover delle etichette dei fluorocromi degli antigeni non co-espressi è sicuro
e non influisce sulla sensibilità e la risoluzione per gli antigeni modulati o scarsamente
espressi.
Bisogna però tener presente che, lo spillover in un antigene co-espresso è ammissibile
se l'obiettivo è solo quello di identificare le cellule brillantemente positive o di
discriminare tra espressione luminosa e medio-forte/tenue (Figura 10).
Figura 10. Perdita di sensibilità quando l’espressione è tenue o negativa. Gli eventi positivi non possono essere
determinati.
Lo spillover, quindi, non può essere ignorato quando l’obiettivo è quello di
discriminare solo tra espressione tenue e negativa, poiché offuscherà i dati,
determinando falsi positivi.
162 Proliferazione cellulare mediante citometria a flusso
ll ciclo cellulare è un processo geneticamente controllato, costituito da una serie
di eventi coordinati e dipendenti tra loro, dai quali dipende la corretta
proliferazione delle cellule eucariotiche. Quest’ultimo si divide in più fasi:
Fase M: Avviene la divisione del nucleo, chiamata mitosi e la separazione
delle due cellule figlie, chiamata citocinesi.
Fase G1 e G2: La cellula continua a crescere ed effettua un monitoraggio
dell’ambiente interno ed esterno per accertarsi che le condizioni siano
adatte per la duplicazione.
Le fasi G sono necessarie per la crescita cellulare e la duplicazione degli
organelli citoplasmatici.
Fase S: La cellula replica il suo DNA nucleare.
La regolazione della proliferazione cellulare è fondamentale per la morfogenesi dei
tessuti durante lo sviluppo degli organismi multicellulari. La perdita del controllo della
proliferazione è alla base di patologie e di malattie quali il cancro; è quindi di vitale
importanza identificare quali cellule vengono duplicate e quali non.
Il monitoraggio del contenuto di DNA nelle cellule può essere facilmente quantificato
mediante citometria a flusso; vedremo di seguito come.
2.1 Danno da radiazione al DNA
Quando le radiazioni ionizzanti interagiscono con un organismo vivente cedono
totalmente o in parte la loro energia alle cellule che lo compongono, provocando la
ionizzazione di atomi o molecole. Il danno biologico, provocato dalla radiazione,
deriva dalla ionizzazione degli atomi che compongono le strutture molecolari alla base
delle cellule negli organismi viventi. Un atomo ionizzato tenderà a produrre nuovi
legami chimici all’interno della molecola alla quale appartiene e come conseguenza
potrà provocare una compromissione delle funzioni vitali della cellula.
I fenomeni fisici con cui le radiazioni interagiscono a livello atomico con la materia
vivente sono le eccitazioni e le ionizzazioni [10, 11]. Tali interazioni fisico-chimiche
sono la causa degli effetti biologici che si manifestano nelle cellule e nei tessuti
irraggiati.
Le radiazioni ionizzanti possono interagire direttamente o indirettamente con il DNA
all’interno della cellula. Nel primo caso la radiazione può interagire direttamente con
gli atomi del bersaglio, i quali vengono eccitati o ionizzati e daranno quindi luogo a
17una serie di eventi che comporterà un danno biologico. Nel secondo caso, invece, la
radiazione interagisce con l’acqua intracellulare formando radicali liberi (Figura 11).
Il DNA è il bersaglio principale dell’effetto biologico delle radiazioni e il danno
procuratogli consiste principalmente in rotture a singolo (Single-Strand Breaks, SSB)
o doppio filamento (Double-Strand Breaks, DSB) dello scheletro zucchero-fosfato.
Il DNA è un polimero costituito da una doppia elica, le cui catene sono composte da
fosfati alternati con gruppi pentosi a ciascuno dei quali è associata una base azotata;
le due catene sono unite insieme da ponti di idrogeno. Le basi azotate sono quattro:
Adenina e Timina che si legano tra di loro mediante un doppio legame idrogeno,
Citosina e Guanina che si legano mediante un triplo legame idrogeno. Queste basi
azotate, raggruppate in gruppi di tre (triplette), sono organizzate in una catena di
nucleotidi che formano i vari geni, localizzati sui cromosomi. Ciascun gene provvede
alla codifica di una particolare proteina. Ogni tripletta determina l’inserimento nella
proteina in formazione di un ben definito amminoacido.
Gli eventuali errori che si possono creare nella codificazione genetica in seguito alle
radiazioni possono dare origine a molecole di DNA che hanno subito dei cambiamenti
di posizione delle triplette con conseguenze sulla proteina costruita. Tali cambiamenti
possono portare a malattie di origine genetica.
L’alterazione strutturale radio indotta degli acidi nucleici comporta un effetto letale a
carico della cellula quando determina la perdita della sua capacità di divedersi. Ciò si
verifica per una compromissione irreparabile dell’informazione ereditaria codificata
nella sequenza dei nucleotidi del DNA.
Figura 11. Catena di eventi che determina il danno biologico delle radiazioni ionizzanti. L’interazione fisica
dura pochi millisecondi; gli effetti biologici si possono manifestare dopo decenni nel singolo individuo e nella
prole [12].
18Di particolare importanza sono gli effetti clinici delle radiazioni ionizzanti e, di
conseguenza, le applicazioni della citometria a flusso sulla microbiologia chimica.
Gli effetti clinici delle radiazioni ionizzanti sono suddivisi in due categorie: danni
deterministici (o non causali), e danni stocastici. I primi, come eritemi cutanei, necrosi
della pelle e così via, riguardano l’individuo esposto a dosi di radiazione modeste-alte.
I secondi, hanno natura probabilistica e si verificano in seguito a mutazione di cellule
somatiche e germinali. In caso di mutazione di cellule somatiche, si verificano
fenomeni quali leucemie e tumori solidi. Il danno alle cellule germinali, invece, può
introdurre una mutazione genetica trasmissibile alla progenie.
Gli effetti stocastici possono manifestarsi dopo anni dall’esposizione e non richiedono
una dose di sogli per la loro comparsa. La loro frequenza di insorgenza è proporzionale
alla dose assorbita, mentre l’entità del danno è indipendente dalla dose; essi possono
essere causati anche dalle procedure di radiologia interventistica, quando il paziente
viene esposto a radiazioni per lunghi periodi di tempo.
In radioprotezione, per esposizioni alle basse dosi di radiazioni ionizzanti, la
valutazione di rischio è basata sulla premessa che qualunque dose di radiazione, non
importa quanto piccola, può risultare in effetti negativa, mediante una relazione
“lineare senza soglia” (Linear No-Threshold cancer risk model, LNT). Tale modello è
riconosciuto e adottato da tutti gli Organismi internazionali che si occupano di
radioprotezione, quali la Commissione sugli effetti biologici delle radiazioni ionizzanti
(BEIR) della National Academy of Science [13].
2.2 Modifiche apportate dalla radiazione sul ciclo cellulare
È stato dimostrato che l’irraggiamento di un’ampia gamma di cellule eucariotiche
induce un rallentamento del ciclo cellulare delle stesse. Tale fenomeno ha diversi
effetti, i quali possono condurre ritardi nella fase G1, S e G2 del ciclo cellulare.
I primi studi sui ritardi del ciclo cellulare indotti dalle radiazioni sono stati condotti su
cellule HeLa, una linea cellulare di adenocarcinoma cervicale umano.
Successivamente all’irraggiamento, tale linea cellulare ha mostrato un ritardo di
transizione provvisorio, con un conseguente accumulo di cellule in fase S [14]. Per
basse dosi di radiazioni, non è stato osservato alcun ritardo nella fase S, mentre si è
notato un aumento della durata della fase G2 dipendente dalla dose. In generale, si è
osservato un ritardo minimo per le cellule irraggiate nella fase G1 ed un ritardo
maggiore per le cellule irraggiate nella fase S e G2.
Si ritiene che il ritardo nella fase G1, come risposta all’irraggiamento, sia dovuto a un
segnale trasmesso attraverso la proteina soppressore del tumore p53 in risposta al
19danno cellulare. Kastan et al. [15] hanno mostrato che l’irraggiamento di cellule di
leucemia mieloblastica ML-I, con dosi tra 0,5 e4 Gy, può causare un accumulo di
cellule sia in G1 che in G2; l’arresto in G1 è predominante per basse dosi, mentre quello
in G2 per alte dosi.
Le proteine p53 agiscono come attivatore trascrizionale causando una maggiore
sintesi di proteine regolatrici implicate nell’arresto del ciclo cellulare (Figura 12).
L’aumentata espressione di p53, dovuta al danno del DNA, porta all’induzione di una
proteina chiamata WAF1\Cip1. Quest’ultima inibisce la fosforilazione dell’istone HI da
parte di ciclina\Cdk2 e complessi di ciclina\Cdk4 in vitro [16].
Figura 12. Rappresentazione schematica di punti potenziali del ciclo cellulare in cui il WAF1\Cip1 indotto da
p53 può inibire la progressione del ciclo cellulare in risposta ai danni del DNA o segnali di arresto della crescita.
L’arresto del ciclo cellulare risulta dall’inibizione dell’attività della ciclina\Cdk chinasi [17].
Per quanto riguarda la fase S, invece, il ritardo nella progressione del ciclo cellulare è
dovuto al rallentamento della velocità di sintesi del DNA. La risposta alla dose di
questo effetto è bifasica, riflettendo la presenza di componenti sia radiosensibili
(bassa dose) che radioresistenti (alta dose).
La componente radioresistente è dovuta a una riduzione della velocità di
allungamento della catena del DNA [18], mentre la componente radiosensibile è
dovuta a un ritardo dell’inizio del replicone [19].
L’arresto delle cellule eucariotiche in G2, dopo l’irraggiamento, sembra essere un
fenomeno universale che, a differenza dell’arresto di G1, non è influenzato dallo stato
del gene p53. L’entità dell’arresto di G2 può essere influenzata dall’espressione di altri
geni oncologici.
Per studiare meglio tale ritardo, sono state analizzate linee di fibroblasti di embrioni
di ratto radiosensibili (REF). Si è dimostrato che tali linee cellulari mostrano differenti
20tempi di ritardo G2 rispetto ad altre linee di cellule dello stesso tipo radioresistenti
[17].
I risultati di tale studio hanno suggerito che le differenze osservate nella
radiosensibilità nelle cellule REF devono essere dovute a fattori come le differenze
nella progressione del ciclo cellulare tra linee cellulari sensibili e resistenti alle
radiazioni.
Si è dimostrato, inoltre, di poter ridurre notevolmente il ritardo G2 indotto dalle
radiazioni nelle cellule HeLa utilizzando staurosporina o caffeina.
Gli studi che utilizzano la caffeina hanno fornito ulteriore supporto per il ruolo del
ritardo G2 nelle cellule irradiate sopravvissute; questa sostanza, infatti, riduce o
abolisce il ritardo G2 indotto dalle radiazioni e rende le cellule più sensibili
all'irraggiamento [20].
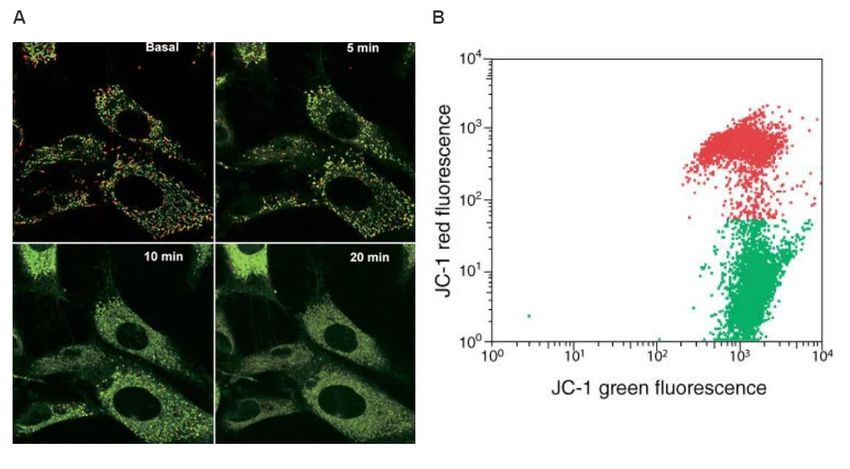
2.3 Misurazione della sintesi del DNA
La sintesi del DNA è un processo alla base dell’eredità biologica e si verifica in tutti gli
organismi viventi.
Tale processo avviene mediante replicazioni semi conservativa: si producono due
repliche identiche di DNA da una molecola di DNA originale.
Tra gli approcci per valutare la proliferazione cellulare mediante citometria a flusso,
troviamo la misurazione della sintesi del DNA con 5-etinil-2'-deossiuridina (EdU) o
colorazione con bromodeossiuridina (BrdU).
Il BrdU è un nucleoside analogo alla timina e può essere incorporato nel DNA appena
sintetizzato.
Nel test BrdU, le cellule che proliferano nell’intervallo temporale in cui è presente
l’analogo temporale della timina, incorporeranno BrdU nel DNA di nuova sintesi.
Queste cellule proliferanti possono essere identificate utilizzando un protocollo di
colorazione BrdU basato su anticorpi, i quali sono marcati con sonde fluorescenti e
riconoscono l’analogo (Figura 13).
21Figura 13. Fasi di incorporazione del BrdU all’interno del DNA delle cellule in analisi (destra) ed esempio di
protocollo utilizzato per questo tipo di test (sinistra) [21].
Per il rilevamento di BrdU, che è incorporato nel DNA appena sintetizzato, è
necessaria la denaturazione dello stesso (Figura 13). Il trattamento con acido, calore
e nucleasi sono opzioni per la denaturazione. Questi reagenti, però, non solo possono
indurre artefatti indesiderati a causa del forte stress al quale sottopongono il
materiale cellulare, ma estendono anche il tempo di rilevamento; tali trattamenti
possono inoltre causare colorazioni incoerenti e diminuzione del segnale di
colorazione.
Per quanto riguarda, invece, il test EdU, questo non è basato sugli anticorpi e quindi
non richiede la denaturazione del DNA per il rilevamento del nucleoside incorporato.
L’EdU è, anch’egli, un nucleoside analogo alla timina e viene incorporato all’interno
del DNA cellulare con tecniche di chimica “a scatto” (click chemistry) [22], basate cioè
sulla possibilità di sintetizzare sostanze complesse in modo semplice e rapido, unendo
molecole più piccole. Dopo aver incorporato l’EdU nel DNA, le cellule marcate con il
reagente contenente EdU vengono misurate rispetto al DNA totale contenuto in una
cellula colorata, ad esempio, con PI (ioduro di propidio), ovvero un agente
fluorescente usato per colorare cellule o acidi nucleici.
22Figura 14. Fasi di incorporazione del EdU all’interno del DNA delle cellule in analisi (destra) ed esempio di
protocollo utilizzato per questo tipo di test (sinistra) [21].
L’EdU, incorporato nel DNA appena sintetizzato, viene quindi rilevato senza alcuna
necessità di denaturazione (Figura 14). In figura 15 è riportato un esempio di misura
delle frazioni di cellule nelle diverse fasi del ciclo cellulare effettuato su cellule
staminali embrionali di ratto [23]; in figura (pannello a sinistra) è chiaramente visibile
la differenza in termini di espressione del fluorocromo legato alla presenza dell’EdU
per le cellule nella fase S, nella quale avviene la replicazione del DNA.
Figura 15. Esempio di quantificazione delle diverse fasi di ciclo cellulare per cellule staminali embrionali di
ratto (ES) marcate con PI ed EdU effettuato con citometro a flusso. Sono riportate i valori ottenuti in termini
di segnale di fluorescenza per i due diversi fluorocromi (a sinistra) e le percentuali di cellule ottenute per le
diverse fasi cellulari (a destra) [23].
232.4 Monitoraggio di generazioni di divisione cellulare
Con il monitoraggio di generazione di divisione cellulare ci si riferisce alla tecnica che
permette di etichettare permanentemente alcune cellule con marcatori fluorescenti,
senza cambiarne la morfologia, per poi tracciarne le successive generazioni o divisioni
in vivo o in vitro.
A tal scopo si utilizza un estere non fluorescente permanente di una molecola
fluorescente ammino-reattiva, il quale entra nella cellula per diffusione tramite
membrana plasmatica.
La molecola non fluorescente, all’ingresso della cellula, viene quindi convertita, grazie
alle esterasi cellulari, in un derivato fluorescente. Successivamente, l’estere attivo
tende a legarsi covalentemente ai gruppi amminici delle proteine, causando
ritenzione del colorante a lungo termine internamente alla molecola.
Tramite successive divisioni cellulari, le cellule figlie assorbono quasi la metà
dell’etichettatura fluorescente delle loro cellule madri, consentendo così l'analisi
delle intensità di fluorescenza delle cellule etichettate e coltivate in vivo (Figura 16).
Figura 16. Esempio di come le divisioni cellulari successive assorbono l’etichettatura fluorescente delle cellule
madri.
Per riuscire a determinare il numero di generazioni attraverso le quali una cellula è
progredita da quando è stata applicata l’etichetta, l’analisi del livello di fluorescenza
tramite citometria a flusso si dimostra quindi una tecnica efficiente.
Un kit utilizzabile con la nostra strumentazione per tale metodo è il kit CellTrace™
CFSE (ThermoFisher Scientific, Waltham, MA, USA); in figura 17 è riportato un
esempio di risultati di analisi per questo tipo di test utilizzando il nostro strumento.
24Figura 17. Segnale in fluorescenza rilevato da linfociti T umani colorati con il reagente CellTrace ™ CFSE per
5 giorni. I picchi discreti in questi istogrammi rappresentano le generazioni successive di cellule vive, mentre
la generazione genitoriale non stimolata è indicata in blu. L'analisi è stata completata utilizzando un
citometro a focalizzazione acustica Attune™ NxT con eccitazione a 488 nm e un filtro di emissione passa-
banda a 530/30 nm.
253 Saggi di apoptosi per citometria a flusso
Con il termine “apoptosi” si intende uno dei possibili processi di morte cellulare.
Quest’ultimo è di grande importanza in quanto, tramite esso, l’organismo riesce a
liberarsi da cellule e tessuti non necessari. Inoltre, si minimizzano i danni
all’organismo distruggendo sia cellule in eccesso del sistema immunitario, sia cellule
infettate da virus.
Esistono due diversi tipi di morte cellulare: morte cellulare programmata (apoptosi) e
morte cellulare per lesione (necrosi).
La necrosi è una morte passiva determinata da un esaurimento del nutrimento,
danneggiamento della membrana cellulare e rilascio di enzimi che portano allo
sviluppo dell’infiammazione. Durante la necrosi la cellula perde la selettività ionica
del sistema, la quale determina il rigonfiamento del citoplasma e degli organelli
cellulari con conseguente perdita della loro organizzazione strutturale. Le alterazioni
a livello nucleare sono più tardive e si esprimono dapprima con la comparsa di picnosi
(nucleo più piccolo, con cromatina addensata) seguita dalla frammentazione in zolle
della cromatina ed infine si verifica la scomparsa del nucleo stesso per espulsione o
dissoluzione nel citoplasma. La conseguenza di questa dissoluzione nucleare e di
questo rigonfiamento è la lisi cellulare, con rilascio nei tessuti del contenuto
intracellulare. Il materiale cellulare così disperso nell’area circostante determina una
risposta infiammatoria (Figura 18) [24].
L’apoptosi, al contrario della necrosi, determina un danno minimo delle cellule e dei
tessuti circostanti, in quanto tale fenomeno non provoca una risposta infiammatoria
perché non si ha versamento del materiale citoplasmatico nell’ambiente
extracellulare. Infatti, a differenza della cellula necrotica, quella apoptotica perde
rapidamente volume, si stacca dalle cellule vicine perdendo i contatti sia con le cellule
circostanti che con la matrice extracellulare.
Figura 18. Morfologia della cellula nel corso del processo di morte cellulare [24].
26Dal punto di vista morfologico, durante l’apoptosi la cellula mantiene la propria
organizzazione interna. A livello nucleare, invece, si osserva la disgregazione del
nucleolo e la condensazione ed il taglio della cromatina in frammenti. Nelle fasi più
tardive dell’apoptosi si formano delle vescicole sulla superficie cellulare che si
staccheranno dando origine ai corpi apoptotici che verranno fagocitati dai macrofagi
e che contengono organelli, porzioni del citoplasma e della cromatina frammentata
(Figura 18) [24].
Le cascate di morte cellulare sono complesse e dinamiche. Per tale motivo, esistono
tanti saggi di apoptosi diversi per la citometria a flusso.
Tutto ciò evidenzia l’importanza di un approccio parametrico alla valutazione
dell’apoptosi. Inoltre, per il rivelamento di quest’ultima, si preferisce utilizzare una
combinazione di tecniche diverse, in quanto non esiste un singolo parametro in grado
di caratterizzare la morte cellulare.
3.1 Danno da radiazione e apoptosi
Come già introdotto nella sezione 2.1 il danno al DNA indotto dalle RI può risultare
nell’attivazione di un meccanismo di risposta, presente in tutti gli eucarioti, che
include l'induzione della riparazione del DNA accoppiata con un
rallentamento/arresto delle diverse fasi del ciclo cellulare [25, 26]. Se la riparazione
ha esito positivo, la cellula riprende la proliferazione, altrimenti possono innescarsi
ulteriori meccanismi di risposta allo stress genotossico, coinvolgendo l'attivazione di
proteine p53. L'accumulo di p53 modula la trascrizione di molti geni responsivi, che
determinano, a seconda del tipo cellulare e della gravità del danno, l'arresto della
crescita temporaneo o permanente o apoptosi [27-29].
I livelli intracellulari e l’attivazione della proteina sono regolati da modificazioni post-
traduzionali come fosforilazioni, sumoilazioni e acetilazioni [30]. In assenza di stress i
livelli di p53 sono mantenuti bassi e stazionari.
Il fatto che esistano risposte alternative alle RI (Figura 19) indica che debbano esistere
meccanismi di controllo che “decidono” tra le opzioni in base al tipo cellulare, allo
stato della cellula, alla severità del danno e alle condizioni ambientali.
La decisione tra apoptosi e arresto della crescita sembra essere ampiamente
determinata da p53. Per alcune tipologie cellulari non sembra in realtà esserci scelta
e la risposta è predeterminata. Cellule di origine emopoietica o cellule embrionali
precoci rispondono alle RI solo andando in apoptosi. D’altra parte, i fibroblasti non
vanno incontro ad apoptosi anche a dosi elevate di RI ma presentano un arresto
prolungato della crescita. Vi sono però molti altri sistemi cellulari per i quali la scelta
tra apoptosi e blocco esiste: la decisione dipende dalle condizioni di crescita e dalla
gravità del danno [31].
27Sono stati proposti diversi modelli per spiegare come le cellule possano scegliere tra
morte e blocco del ciclo in risposta all’attivazione di p53. Secondo un primo modello
la decisione dipende dalla quantità di p53 attivata e dalla durata della sua attivazione:
più è sostenuta e prolungata l’attivazione di p53 più è probabile che la cellula vada in
apoptosi [32].
Per un secondo modello è più probabile che tipi cellulari con più geni pro-apoptotici
potenzialmente attivi vadano in apoptosi piuttosto che in arresto della crescita
cellulare [33]. Secondo un ulteriore modello la scelta tra apoptosi e blocco del ciclo
dipenderebbe dalla disponibilità di co-fattori che regolerebbero la capacità di p53 di
legare un sottogruppo specifico di geni bersaglio piuttosto che un altro. Tutti i
meccanismi citati sono supportati da dati sperimentali ottenuti in modelli diversi e
sembra pertanto che tutti intervengano nella regolazione della risposta p53- mediata
al danno da RI [34].
Le cellule che sfuggono all’apoptosi dopo danno da RI esteso e irreparabile hanno due
opzioni: andare in catastrofe mitotica quindi morire o rimanere vive, ma in arresto
irreversibile della crescita (Figura 19).
Così come l’apoptosi p53-dipendente avviene solo in alcuni tessuti specifici, la
suscettibilità dei tumori all’apoptosi potrebbe dipendere dal loro tessuto di origine.
Tale ipotesi è stata supportata da uno studio secondo il quale i trattamenti
antineoplastici con chemioterapici o RI possono indurre apoptosi nel tumore solo se
questo origina da tessuti suscettibili all’apoptosi p53 dipendente [35]. Studi clinici
indicano che la morte per apoptosi in risposta al trattamento antineoplastico è
caratteristica di tumori che originano dal tessuto emopoietico e riproduttivo noti per
essere naturalmente proni all’apoptosi. In conclusione, sebbene l’apoptosi possa
essere un fattore importante nel determinare la suscettibilità dei tumori ai
trattamenti antineoplastici negli stadi più precoci della progressione tumorale, essa
ha un ruolo modesto nella maggior parte dei tumori quando questi perdono i loro
meccanismi pro-apoptotici [36].
28Figura 19. Destino della cellula in seguito a RI [35].
3.2 Colorazione dell’annessina V
Un metodo comune per rilevare le cellule apoptotiche è la colorazione mediante
annessina V. Quest’ultima, è una proteina che lega alcuni fosfolipidi chiamati
fosfatidilserine (PS), che normalmente si trovano solo nel foglietto interno rivolto
verso il citoplasma della membrana di una cellula, ma che si capovolgono al foglietto
esterno durante le prime fasi dell'apoptosi [37].
La colorazione dell'annessina V per rilevare le cellule apoptotiche può essere eseguita
solo su cellule e tessuti vivi e riesce a distinguere le diverse vie della morte cellulare:
apoptosi e necrosi.
Durante l’apoptosi vengono attivate le caspasi citosoliche, enzimi che a loro volta
attivano un enzima legato alla membrana chiamato scramblasi. Quest’ultimo
"strapazza" la membrana cellulare traslocando i residui di PS dal lato citoplasmatico
alla superficie esterna. Al contrario, durante la necrosi i residui di PS rimangono dove
sono, ma la membrana stessa si rompe. La colorazione mediante annessina V
capitalizza tali differenze nelle alterazioni di membrana dei due tipi di morte cellulare.
L’annessina V coniugata con un colorante fluorescente, è un ligando naturale
impermeabile alla membrana per PS, e quindi identifica facilmente le cellule in uno
stadio precoce di apoptosi legandosi alla PS esternalizzata.
Va però prestata particolare attenzione all’insorgere di un problema: i falsi positivi.
Tale problema si verifica quando, a seguito della rottura della membrana, l’annessina
V può anche legarsi alla PS sulla faccia interna.
Per ovviare a tale problematica, si utilizza una molecola di colorante, lo ioduro di
propidio (PI) che lega il DNA, che è impermeabile alla membrana e che può entrare in
una cellula solo quando la membrana presenta delle lesioni.
Di conseguenza, se una cellula è solo lieve colorazione di annessina, è apoptotica
precoce, mentre una cellula che è sia PI- che annessina-positiva potrebbe essere
necrotica o apoptotica tardiva [37].
29Per quanto concerne, invece, il processo di colorazione e analisi di morte cellulare, in
primo luogo le cellule vengono raccolte e centrifugate a bassa velocità per prevenire
qualsiasi danneggiamento morfologico. Dopo un'altra fase di centrifugazione, le
cellule vengono raccolte in una soluzione tampone di annessina V contenente calcio,
che è necessario per il legame dell'annessina alla PS. L'annessina V coniugata con FITC
viene quindi aggiunta alla sospensione cellulare, che viene incubata a temperatura
ambiente al buio per consentire l'associazione di annessina e PS. Successivamente,
alla sospensione cellulare già marcata con annessina, viene aggiunto il PI e viene
nuovamente incubata in assenza di luce. Dopo l'incubazione, le cellule vengono lavate
in soluzione salina (Phosphate-buffered saline, PBS), centrifugate e conservate a 0°
gradi fino al momento della misura [37].
Mediante citometria a flusso viene analizzata l’attività di fluorescenza delle cellule.
Dopo la calibrazione della fluorescenza, utilizzando cellule colorate separatamente
con ciascun marcatore (si veda sezione 1.4), vengono acquisiti i dati delle cellule a
doppia colorazione. I segnali di fluorescenza provenienti dalla popolazione cellulare
vengono utilizzati per creare un grafico in cui l'intensità FITC legata all'annessina viene
tracciata sull'asse X e PI sull'asse Y. In figura 20 è riportato un esempio di tale tecnica
eseguita su cellule di Jurkat (leucemia a cellule T, umana) nelle quali l’apoptosi è stata
indotta tramite l’esposizione a camptotecina [38]; le cellule sono state marcate con
annessina V, coniugata con fluorocromo Alexa Fluor 488 (un fluorocromo derivato dal
FITC ma caratterizzato da maggiore stabilità) per identificare le cellule apoptotiche e
con PI per discriminare le cellule morte da quelle vitali. Nel grafico a destra sono
riportate cellule di Jurkat trattate con 10 μM di camptotecina per 4 ore, mentre in
quello di sinistra sono riportati i campioni di controllo non trattati.
Come si può vedere nell’immagine gli spot che sono annessina-Alexa positivi e PI-
negativi si raggruppano sul lato inferiore destro dei grafici, e corrispondono a cellule
apoptotiche precoci, mentre quelli doppi positivi per annessina-Alexa e PI si trovano
in alto a destra, e corrispondono alle cellule apoptotiche o necrotiche tardive. Le
cellule che rimangono non marcate o marcate in modo non significativo sia per
l'annessina che per la PI sono cellule “vitali”.
30Puoi anche leggere

















































