IV Corso di aggiornamento sui farmaci: Expanded access, off label e farmacovigilanza
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
IV Corso di aggiornamento sui farmaci:
Expanded access, off label e farmacovigilanza
“Il decreto 8 Maggio 2003 “Expanded Access”: esperienze della sua applicazione”
Maria Federica Barchetti
Auditorium del Ministero della Salute
Roma 26 Gennaio 2011
Agenzia Italiana del Farmaco – Sperimentazione Clinica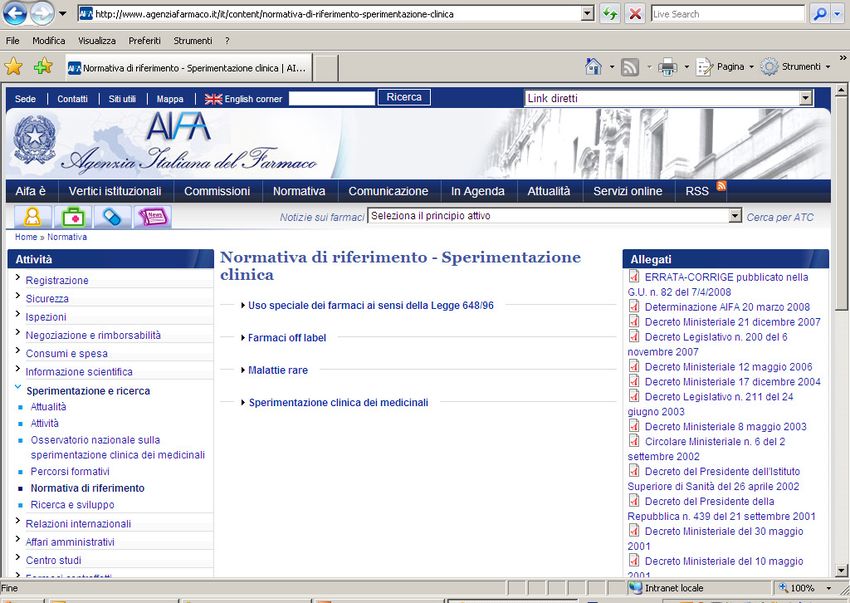
Agenda
Impegno istituzionale dell’AIFA
DM 08/05/2003 “Uso terapeutico di medicinale
sottoposto a sperimentazione clinica c.d. uso
compassionevole
Contesto normativo e linee guida europee
Riflessioni Etiche
Agenzia Italiana del Farmaco – Sperimentazione Clinica
Legge n. 326/2003 Agenzia Italiana del Farmaco – Sperimentazione Clinica

Agenzia Italiana del Farmaco – Sperimentazione Clinica
Usi speciali dei medicinali
I riferimenti normativi
Legge n. 648 del 23 Dicembre 1996
Legge n. 94 8/4/1998
DM 8/5/2003 G.U. 173 del 28 Luglio 2003
Agenzia Italiana del Farmaco - Sperimentazione Clinica
Agenzia Italiana del Farmaco – Sperimentazione Clinica

Nicchie terapeutiche
L’attività del medico non si esaurisce all’ordinaria prescrizione di
specialità medicinali nelle indicazioni, vie o modalità di somministrazione
previste dall’autorizzazione all’immissione in commercio.
Esistono aree terapeutiche così dette di “nicchia” che riguardano
bisogni clinici insoddisfatti, needs medical unmet, per il trattamento di
malattie rare
pazienti in condizioni gravi con scarse opzioni terapeutiche
pazienti in situazioni pericolo di vita
pazienti non responder alle terapie standard
popolazioni di pazienti poco studiate nei clinical trials
Agenzia Italiana del Farmaco – Sperimentazione Clinica
DM 8/5/2003
G.U. 173 del 28 Luglio 2003
Uso terapeutico di medicinale sottoposto a
sperimentazione clinica
c.d. uso compassionevole
Agenzia Italiana del Farmaco - Sperimentazione Clinica
DM 8/05/2003
E’ lo strumento normativo che in
Italia definisce le procedure operative da
seguire per consentire l’accesso a terapie
farmacologiche sperimentali attraverso
protocolli così definiti di accesso allargato
o “expanded access”.
Agenzia Italiana del Farmaco - Sperimentazione Clinica
“Uso terapeutico di medicinale sottoposto a sperimentazione clinica”
Art.1
1. Un medicinale prodotto in stabilimento farmaceutico,
autorizzato ai sensi dell’articolo 3 del DM 11 Febbraio 1997
sottoposto a sperimentazione clinica sul territorio italiano o in
Paese estero, privo dell’autorizzazione di cui all’art. 8 del
decreto legislativo del 29 maggio 1991, n.178, può essere
richiesto all’impresa produttrice per uso al di fuori della
sperimentazione clinica quando non esista valida alternativa
terapeutica al trattamento di
patologie gravi
malattie rare
condizioni di malattia che pongono il paziente in
pericolo di vita.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaD.Lvo n. 211/2003 art. 2 comma 1 d) d) “medicinale sperimentale”; una forma farmaceutica di un principio attivo di un placebo saggiato come controllo in una sperimentazione clinica compresi i prodotti che hanno ottenuto un’autorizzazione di commercializzazione ma che sono utilizzati o preparati (secondo formula magistrale o confezionati) in forme diverse da quella autorizzata, o quando sono utilizzati per indicazioni non autorizzate o per ottenere ulteriori informazioni sulla forma autorizzata Agenzia Italiana del Farmaco – Sperimentazione Clinica
art. 2 comma 1 DM 08/05/2003
La richiesta può essere autorizzata alle seguenti
condizioni:
a) il medicinale sia richiesto nella medesima
specifica indicazione terapeutica, di studi clinici
sperimentali, in corso o conclusi, di fase III o, in casi
particolari di condizioni di malattia che pongano il
paziente in pericolo di vita, di studi clinici già conclusi di
fase II;
b) i dati disponibili sulle sperimentazioni siano
sufficienti a formulare un favorevole giudizio
sull’efficacia e la tollerabilità del medicinale richiesto
Agenzia Italiana del Farmaco – Sperimentazione ClinicaREGISTRAZIONE
Ricerca e
Ricerca CLINICA POST MARKETING
sviluppo
e svilupo registrativo surveillance
PRE CLINICO
FASE I
Volontari sani : sicurezza, effetti
biologici, metabolismo, farmacocinetica
• PRESCRIZIONE
• SEGNALAZIONE REAZIONI
AVVERSE
FASE II • FARMACOVIGILANZA,
FARMACOSORVEGLIANZA
Campione di pazienti : sicurezza, effetti • FARMACOEPIDEMIOLOGIA
biologici, metabolismo, farmacocinetica • STUDI DI MORTALITA’
• POPOLAZIONE
FASE III
Campione più ampio di pazienti
selezionati : sicurezza ed efficacia
BREVE TERMINE
LUNGO TERMINE FASE IV
Studi nell’ animale : tossicità cronica, effetti
sulla riproduzione, teratogenicità
1- 3 2-10
anni anni
Agenzia Italiana del Farmaco – Sperimentazione Clinicaart. 3 DM 08/05/2003
La fornitura del medicinale potrà essere richiesta alla ditta
produttrice:
a) dal medico per uso nominale nel singolo paziente non trattato
nell’ambito di studi clinici
b) da più medici operanti in diversi centri o da gruppi
collaborativi multicentrici
c) dai medici o da gruppi collaborativi, per pazienti che hanno
partecipato a una sperimentazione clinica che ha dimostrato un
profilo di efficacia e tollerabilià tale da configurare la necessità,
per coloro che hanno partecipato al trial, a fruire con la
massima tempestività dei suoi risultati.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaArt. 4 DM 08/05/2003
1. A seguito della richiesta, l'impresa produttrice può fornire il
farmaco sulla base di un protocollo in cui siano presenti ed
adeguatamente documentate:
a) la motivazione clinica della richiesta;
b) i dati pertinenti relativi alla efficacia ed alla tollerabilità;
c) il grado di comparabilità dei pazienti inclusi nelle
sperimentazioni di cui all’articolo 2, comma 1, lettera a), e di
coloro per cui è formulata la richiesta;
d) le modalità di informazione al paziente;
e) le modalità di raccolta dati, secondo la logica di uno studio
osservazionale.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaArt. 4 DM 08/05/2003
2. Il protocollo deve essere:
a) sottoposto da parte del medico alla approvazione da parte del
Comitato Etico nel cui ambito di competenze origina la
richiesta, il quale può operare anche mediante procedura di
urgenza, accompagnato da una nota di assunzione di
responsabilità del trattamento secondo protocollo da parte del
medico richiedente;
b) notificato, contestualmente alla notifica di cui alla lettera a), al
Ministero della Salute – Direzione generale valutazione
medicinali e farmacovigilanza – Ufficio Sperimentazione clinica
- i cui uffici possono formulare un eventuale giudizio
sospensivo della procedura o dell'uso.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaComitato Etico
Esprimere parere
1. Idoneità del medico richiedente
2. Protocollo d’uso del medicinale, IB
3. Documento di informazione al paziente e il modulo di
acquisizione del consenso informato
4. Valutazione del rapporto rischio/beneficio del medicinale
5. Paziente candidato al trattamento
Un parere favorevole autorizza la fornitura del medicinale
Un parere non favorevole la respinge
Agenzia Italiana del Farmaco – Sperimentazione ClinicaArt. 4 DM 08/05/2003
3. Il medicinale è fornito gratuitamente
dall’impresa autorizzata. Per l’eventuale
ingresso del farmaco presso gli uffici
doganali preposti, dovrà essere
presentata l’approvazione da parte
del Comitato Etico competente.
La fornitura del medicinale da parte dell’Azienda Farmaceutica è
subordinata al parere favorevole espresso dal Comitato Etico
Agenzia Italiana del Farmaco – Sperimentazione ClinicaCheck-list di documenti da notificare all’Ufficio Ricerca e
Sperimentazione Clinica dell’AIFA
1. Richiesta del medicinale da parte del medico con assunzione di
responsabilità;
2. Notifica del/dei pazienti candidati al trattamento, breve
motivazione clinica della richiesta
3. Dichiarazione di disponibilità della ditta alla fornitura gratuita del
medicinale
4. Protocollo d’uso del medicinale, RCP
5. Documentazione di informazione al paziente e modulo di
consenso informato;
6. Copia del Parere espresso dal Comitato Etico competente;
Agenzia Italiana del Farmaco – Sperimentazione ClinicaNotifica della documentazione all’Ufficio Sperimentazione Clinica
dell’AIFA
L’Ufficio Sperimentazione Clinica
riceve notifica della documentazione
relativa “all’uso terapeutico” dei
medicinali e ne verifica la conformità
al DM 8/5/2003 e alla normativa
applicabile.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaArticolo 4 5 Qualora l’utilizzazione del medicinale avvenga al di fuori di quanto stabilito all’art. 1 ovvero non vengano rispettati gli adempimenti e le limitazioni stabilite dall’autorizzazione, o qualora, comunque, lo ritenga opportuno per la tutela della salute pubblica, il Ministero della Salute può sospendere o vietare l’ulteriore cessione e l’impiego di medicinali di cui al presente decreto. Agenzia Italiana del Farmaco - Sperimentazione Clinica
Articolo 4 4 I dati relativi all’uso del farmaco, di cui al presente decreto, possono essere utilizzati come supporto, ma non sostitutivi, di quanto richiesto nelle procedure di autorizzazione all’immissione in commercio, ai sensi dell’art. 8 del decreto legislativo del 29 Maggio 1991, n. 178. Agenzia Italiana del Farmaco – Sperimentazione Clinica
Chiusura del programma
Le Ditte Farmaceutiche provvederanno a
comunicare all’Ufficio Sperimentazione Clinica
dell’AIFA e ai centri la data di inizio e la data di
chiusura del programma previsto in ambito
nazionale in corrispondenza della avvenuta
pubblicazione in G.U. della Repubblica Italiana della
Determinazione AIFA che autorizza la
commercializzazione del medicinale sul territorio
nazionale e ne stabilisce prezzo e regime di
rimborsabilità.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaRequisiti di qualità richiesti per un medicinale sperimentale
DM 7 Novembre 2008 G.U. n.80 del 6/4/2009
art. 2 comma 1 a)
IMP risponda ai requisiti di qualità richiesti:
art. 13 del Decreto legislativo n. 211 del 24 Giugno 2003
capo III del decreto legislativo 6 Novembre 2007, n.200 per
la produzione dei medicinali secondo GMP
Annex 13 Norme di Buona Fabbricazione dei Medicinali
Agenzia Italiana del Farmaco – Sperimentazione ClinicaRuolo dell’AIFA –Ufficio Ricerca e Sperimentazione Clinica
Garantire il necessario controllo e la corretta gestione da parte dell’ufficio
sperimentazione clinica dell’AIFA delle notifiche delle richieste di medicinali
formulate ai sensi del DM 08/05/2003
Garantire ai pazienti la migliore qualità possibile dell’assistenza
farmaceutica attraverso l’accesso a medicinali innovativi, a farmaci orfani
per malattie rare, là dove a giudizio del medico non esistano ulteriori
opportunità terapeutiche
Promuovere la tracciabilità delle attività sul territorio nazionale
Intervenire, se necessario, con misure restrittive per motivi di tutela della
salute pubblica
Agenzia Italiana del Farmaco – Sperimentazione ClinicaAgenzia Italiana del Farmaco – Sperimentazione Clinica
Agenzia Italiana del Farmaco – Sperimentazione Clinica
Riferimenti Normativi
D.lvo 211 24 Giugno 2003:
2003 attuazione della direttiva 2001/20/CE relativa
all’applicazione della buona pratica clinica nell’esecuzione delle sperimentazioni
cliniche di medicinali per uso clinico
Direttiva europea 2001/20/CE del 4 Aprile 2001 concernente il
ravvicinamento delle disposizioni legislative, regolamentari ed amministrative
degli Stati Membri relative all’applicazione della buona pratica clinica
nell’esecuzione della sperimentazione clinica di medicinali per uso umano
Decreto Legislativo 24 Aprile 2006 n. 219 Attuazione della Direttiva
2001/83/CE(e successive direttive di modifica) relativa ad un codice comunitario
concernente i medicinali per uso umano,nonché della direttiva 2003/94/CE
DM 11/02/1997 modalità di importazione di medicinali esteri e successive
modifiche
Agenzia Italiana del Farmaco – Sperimentazione ClinicaRiferimenti Normativi
DM 12 Maggio 2006 Requisiti minimi per l’istituzione, l’organizzazione e il
funzionamento dei Comitati Etici per le sperimentazioni cliniche dei medicinali
Decreto Legislativo 6 Dicembre 2007 n. 200 Principi e linee guida dettagliate
per la buona pratica clinica relativa ai medicinali in sperimentazione
DM 15 Luglio 1997,
1997 recepisce le linee guida delle Good Clinical practice,
elaborate in sede ICH (International Coference of Harmonisation e approvate
dall’Emea nel Luglio 1996): “Recepimento delle linee guida dell’Unione europea
di buona pratica clinica per la esecuzione delle sperimentazioni cliniche dei
medicinali
Good Clinical Practice,
Practice, GCP;
GCP è uno standard internazionale di etica e qualità
scientifica per progettare, condurre, registrare e relazionare gli studi clinici che
coinvolgano soggetti umani. L’aderenza a questi standard di GCP garantisce
pubblicamente non solo la tutela dei diritti, della sicurezza e del benessere dei
soggetti che partecipano allo studio, in conformità con i principi stabiliti dalla
Dichiarazione di Helsinki, ma anche l’attendibilità dei dati relativi allo studio
clinico.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaRiferimenti Normativi
Dichiarazione di Helsinki (1964 e successive) della World Medical
Association contenente i principi etici cui i medici di tutto il mondo sono
obbligati ad attenersi per la conduzione delle ricerche cliniche
DM 7 Novembre 2008 recante modifiche ed integrazioni ai decreti 19
Marzo 1998; 8/5/2003 e al DM 12 Maggio 2006;
Regolamento CE n.141/2000 del Parlamento Europeo e del Consiglio
concernente i medicinali orfani.
Il Regolamento n.141/2000 stabilisce le procedure comunitarie per
l’attribuzione della qualifica di medicinale orfano secondo criteri obiettivi
basati su una prevalenza della affezione per la quale è ricercata una
diagnosi, una profilassi, una terapia non superiore a 5 casi su 10000 ;
i medicinali per la cura di una affezione potenzialmente letale,
gravemente invalidante o grave e cronica dovrebbero beneficiare di
incentivi anche qualora la prevalenza sia superiore a 5 casi su 10000
(considerando 5)
Agenzia Italiana del Farmaco – Sperimentazione ClinicaRiferimenti Normativi
Regolamento (CE) N. 726/2004 del Parlamento europeo e del
consiglio del 31 Marzo 2004 che istituisce procedure comunitarie
per l’autorizzazione e la sorveglianza dei medicinali per uso
umano e veterinario e che istituisce l’Agenzia Europea dei
medicinali
Considerando 7
L’esperienza acquisita ha dimostrato la necessità di istituire una procedura
comunitaria centralizzata di autorizzazione obbligatoria dei medicinali ad alta
tecnologia, in particolare quelli derivati dalle biotecnologie, per conservare l’alto
livello di valutazione scientifica di tali medicinali nella comunità e per preservare
di conseguenza la fiducia dei pazienti e degli operatori sanitari in tale valutazione
Agenzia Italiana del Farmaco – Sperimentazione ClinicaRegolamento (CE) N. 726/2004
ALLEGATO
MEDICINALI CHE DEVONO ESSERE AUTORIZZATI DALLA COMUNITÀ
COMUNITÀ
1. Medicinali derivati dai seguenti procedimenti biotecnologici:
— tecnologie da DNA ricombinante,
— espressione controllata di geni portatori di codici per proteine biologicamente attive nei procarioti e negli
eucarioti, comprese cellule trasformate di mammiferi,
— metodi a base di ibridomi e di anticorpi monoclonali.
2. Medicinali veterinari destinati principalmente ad essere utilizzati come stimolatori per migliorare la crescita o la
produttività degli animali trattati.
3. Medicinali per uso umano contenenti una nuova sostanza attiva non autorizzata nella Comunità alla data di
entrata
in vigore del presente regolamento, aventi come indicazione terapeutica il trattamento di una delle seguenti
malattie:
— sindrome da immunodeficienza acquisita;
— cancro;
— disordini neurodegenerativi;
— diabete;
e, con effetto da 20 maggio 2008,
— malattie autoimmuni e altre disfunzioni immunitarie;
— malattie virali.
Dopo 20 maggio 2008 la Commissione, sentita l'agenzia, può presentare proposte appropriate di modifica del
presente punto sulle quali il Consiglio decide a maggioranza qualificata.
4. Medicinali che sono designati come medicinali orfani ai sensi del regolamento (CE) n. 141/2000.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaRegolamento (CE) N. 726/2004
Art. 83
1 In deroga all’art. 6 della direttiva 2001/83/CE, gli Stati membri possono
mettere a disposizione, per uso compassionevole, un medicinale per
uso umano appartenente alle categorie di cui all’art. 3, paragrafi 1 e 2
del presente regolamento.
2 Ai fini del presente articolo, per uso compassionevole si intende la
messa a disposizione, per motivi umanitari, di un medicinale
appartenente alle categorie di cui all’art. 3, paragrafi 1 e 2, ad un
gruppo di pazienti affetti da una malattia cronica o gravemente
invalidante o la cui malattia è considerata potenzialmnente letale, e
che non possono essere curati in modo soddisfacente con un
medicinale autorizzato. Il medicinale in questione deve essere oggetto
di una domanda di autorizzazione all’immissione in commercio a
norma dell’art. 6 del presente regolamento o essere sottoposto a
sperimentazione clinica.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaAgenzia Italiana del Farmaco – Sperimentazione Clinica
Guideline on Compassionate Use of medicinal products
Guideline on compassionate use (CU) of medicinal products, pursuant to
article 83 of regulation (EC) N. 726/2004 Doc.Ref: EMEA/27170/2006
CU programmes are intended to facilitate the availability to patients of new
treatment options under development
CU programmes, making medicinal products available either on a named
patient basis or to cohorts of patients, are governed by individual Member States
(MS) legislation.
CU versus clinical trials : From a methodological point of view, clinical trials
are practically the only means of obtaining reliable and interpretable efficacy and
safety data for a medicinal product. Although safety data may be collected during
CU, such programmes cannot replace clinical trials for investigational purposes
CU is not a substitute for properly conducted trials. CU should not slow down
the implementation or continuation of clinical trials intended to provide essential
information relative to the benefict/risk balance of a medicinal product
Agenzia Italiana del Farmaco – Sperimentazione ClinicaGuideline on Compassionate Use of medicinal products
Guideline on compassionate use of medicinal products,
products, pursuant to article 83 of
regulation(EC)
regulation(EC) N. 726/2004
Compassionate use versus off-
off-label use:
use in this guideline, cu does not refer to use of
an authorised medicinal product for an indication different from the one mentioned in
section 4.1 of the summary of product characteristics (SPC) i.e. off-label use
Pharmacovigilance:
Pharmacovigilance: pharmacovigilance rules and responsabilities are applicable to
medicinal products for which an opinion on the conditions for compassionate use has
been adopted
The EMEA’
EMEA’s role in Compassionate use The EMEA’s recommendations may help
to make compassionate use programmes similar across the European Union. They
may also help to make existing compassionate use programmes more trasparent.
London 19 July 2007 Doc.Ref:
Doc.Ref: EMEA/72144/2006
Agenzia Italiana del Farmaco – Sperimentazione ClinicaEuropean Medicines Agency
What is compassionate use?
Compassionate use is a way of making available to patients with an
unmet medical need a promising medicine which has not yet been authorised
(licensed) for their condition.
A medicine can be marketed in the European Union (EU) only after it has
been authorised. However, it is sometimees in the interest of patients to have
access to medicines before authorisation. Special programmes can be set up to
make these medicines available to them under defined conditions.This is know as
“compassionate use”.
Compassionate use programmes can only be put in place for medicines
that are expected to help patients with life-threating, long-lasting or seriously
disabling illnesses. These programmes are expected to benefict seriously ill patients
who currently cannot be treated satisfactorily with authorised medicines, or who
have a desease for which no medicine has yet been authorised. The compassionate
use route may be a way for patients who cannot enrol in an ongoing clinical trial to
obtain treatment with a potentially life-saving medicine. EMEA/72144/2006 (rev)
Agenzia Italiana del Farmaco – Sperimentazione ClinicaAgenzia Italiana del Farmaco – Sperimentazione Clinica
Agenzia Italiana del Farmaco – Sperimentazione Clinica
Alcune esperienze dell’applicazione del DM 8/5/2003
Agenzia Italiana del Farmaco – Sperimentazione ClinicaMedicinali richiesti ai sensi del DM 08/05/2003
TMC 125 etravirina infezioni da HIV in pazienti Janssen Cilag
NNTRI pluritrattati e/o multiresistenti
MK-0518 raltegravir infezioni da HIV in pazienti MSD
Inibitore integrasi pluritrattati e/o multiresistenti
TMC114 darunavir infezioni da HIV in pazienti Janssen Cilag
IP pluritrattati e/o multiresistenti
Temozolamide Tumori cerebrali Schering Pluogh
gliomi
Panitumumab Carcinoma del colon-retto Amgen
metastatico
Omalizumab Asma allergico moderato- Novartis
grave non controllato da
terapie convenzionali
Agenzia Italiana del Farmaco – Sperimentazione ClinicaMedicinali richiesti ai sensi del DM 08/05/2003
Ipilimumab Melanoma metastatico BMS
Ixabepilone Carcinoma metastatico della BMS
mammella pretrattati e
resistenti a taxani, antracicline
e/o capecitabina
Tamiflu IV Trattamento di pazienti in Roche
condizioni critiche con
infezione influenzale H1N1o
stagionale da virus A o B
Ozurdex Edema maculare Allergan
BRVO-CRVO
Trastuzumab Carcinoma gastrico Roche
metastatico HER 2+
Agenzia Italiana del Farmaco – Sperimentazione ClinicaOrphan Medicinal Products
Trade NameProduct EU Designation Designated Orphan Designation date Sponsor
Indication
Nexavar EU/04/207 Renal cell 29/07/2004 Bayer
Sorafenib EU/3/6/364 carcinoma 11/04/2006
epatocarcinoma
Exjade Treatment of chronic 13/3/2002 Novartis Europharm
Deferasirox EU/3/02/92 iron overload Limited, UK
requiring chelation
therapy
Torisel EU/3/06/365 Treatment of renal 6/4/2006 Wyeth Europa
Temsirolimus cell carcinoma Limited
Prialt EU/3/01/050 Treatment of chronic Eisai Limited
Ziconotide pain requiring 9/7/2001
intraspinal analgesia
Agenzia Italiana del Farmaco – Sperimentazione ClinicaOrphan Medicinal Products
Revatio EU/3/03/178 Treatment of 12/12/2003 Pfizer Ltd
sildenafil pulmonary arterial
hypertension
Nelarabine EU/3/05/293 Treatment of acute 23/12/2005 Glaxo Group
lynphoblastic Ltd
leukaemia
Revlimid
EU/3/03/177 Mieloma Multiplo 12/12/2003 Celgene Europe
Lenalidomide
Diacomit stiripentol EU/3/04/207 Treatment of severe 5/12/2001 Biocodex
myoclonic epipepsy
in infancy
Agenzia Italiana del Farmaco – Sperimentazione ClinicaOrphan Medicinal Products
Sprycel EU/3/05/339 Treatment of chronic 3/12/2005 Bristol-Myers
Dasatinib EU/3/5/338 myeloid leukaemia 23/12/2005 Squibb Pharma
Treatment of acute EEIG
lymphoblastic
leukaemia
Forodesina EU/3/06/428 Linfoma cutaneo a 29/01/2007 Mundipharma
cellule T
Forodesina EU/3/06/421 Acute Lynphoblastic 18/12/2006 Mundipharma
leukaemia
Myozyme EU/3/00/018 Treatment of glycogen 14/02/2001 Genzyme Europe
Recombinant human
Storage disease type II B.V.
acid alpha-
( pompe’s disease)
glucosidase
Agenzia Italiana del Farmaco – Sperimentazione ClinicaOrphan Medicinal Products
Eltrombopag EU/3/07/467 Idiopathic 3/8/2007 GSK
thrombocytopenic
purpura
Afamelanotide EU/3/09/648 Treatment of solar 24/07/2004 Clunivel UK Linited
urticaria
Imatinib EU/3/05/306 Treatment of 26/08/2005 Novartis
mastocytosis
Imatinib EU/3/1/061 Treatment of 20/11/2001 Novartis
GIST
Agenzia Italiana del Farmaco – Sperimentazione ClinicaOrphan Medicinal Products
Pasireotide EU/3/09/671 Cushing’s disease 8/10/2009 Novartis
Givinostat EU/3/09/719 Polytemia vera 3/272010 Italfarmaco
Ofatumumab EU/3/08/581 Cronic lymphocytic 7/11/2008 GSK
leukaemia
Vorinostat EU/3/04/205 Cutaneus T-cell 21/6/205 MSD
Lynphoma
Agenzia Italiana del Farmaco – Sperimentazione ClinicaOrphan Medicinal Products
Muramyl Tripeptide EU/3/04/2006 Osteosarcoma 21/06/2006 AAIFarma
Mifamurtide
Mepolizumab EU/3/04/213 Treatment of 29/07/2004 GSK
Hypereosinophilic
syndrome
Everolimus EU/3/07/449 Treatment of renal 5/6/2007 Novartis
cell carcinoma
Everolimus EU/3/07/488 Grastro-entero- 14/11/2007 Novartis
pancreatic
Neuroendocrine
tumors
Agenzia Italiana del Farmaco – Sperimentazione ClinicaAgenzia Italiana del Farmaco – Sperimentazione Clinica
Agenzia Italiana del Farmaco – Sperimentazione Clinica
DM 08/05/2003
Il DM 08/05/2003 non deve essere
considerato un riferimento normativo per
consentire una sperimentazione clinica
allargata ma uno strumento per favorire
l’accesso precoce, early access, a terapie
farmacologiche sperimentali in assenza di
valide alternative terapeutiche
Agenzia Italiana del Farmaco – Sperimentazione ClinicaDM 8/5/2003
La tipologia di pazienti cui il decreto
si rivolge richiede una particolare
tutela e protezione appartenendo
alla categoria dei soggetti così
definiti “vulnerabili” dalle GCP
(1.61 GCP DM 15/7/1997)
Agenzia Italiana del Farmaco – Sperimentazione ClinicaSoggetti Vulnerabili
1.61 GCP DM 15/7/1997
Individui la cui decisione di offrirsi come volontari in uno studio
clinico può essere influenzata impropriamente dall’aspettativa,
sia essa giustificata o meno, di benefici legati alla
partecipazione, oppure di una possibile azione di ritorsione da
parte di individui gerarchicamente superiori, in caso di rifiuto a
partecipare……………
Altri soggetti vulnerabili possono essere: pazienti affetti da
malattie incurabili………..pazienti in situazioni di
emergenza……………..minori e persone incapaci di dare il
proprio consenso.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaGood Clinical Practice
3.1 GCP Responsabilità
Un IRB/IEC deve tutelare i diritti, la sicurezza e il
benessere di tutti i soggetti che partecipano allo studio.
Deve essere prestata particolare attenzione agli studi che
coinvolgono soggetti vulnerabili
Agenzia Italiana del Farmaco – Sperimentazione ClinicaComitato Etico
Art. 1 DM 12 Maggio 2006
1 Il Comitato Etico per le sperimentazioni
cliniche di medicinali di cui all’art. 2,
comma 1, lettera m) e all’art. 6 del decreto
legislativo 24 Giugno 2003, n.211, è un
organismo indipendente che ha la
responsabilità
responsabilità di garantire la tutela dei
diritti, della sicurezza e del benessere dei
soggetti in sperimentazione e di fornire
pubblica garanzia di tale sicurezza.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaDM 12 Maggio 2006
art. 2 Istituzione e composizione
Due clinici
Un medico di medicina generale territoriale e/o un pediatra di llibera
ibera scelta
Un biostatistico
Un farmacologo
Un farmacista (ex officio) del servizio farmaceutico della istituzione
istituzione di ricovero o
territoriale, sede della sperimentazione clinica dei medicinali; nei casi di cui all’
all’art.
1, comma 2, un farmacista del servizio sanitario regionale
Il Direttore sanitario (ex officio) e ove applicabile, come nel caso degli istituti di
scientifico
ricovero e cura a carattere scientifico, del direttore scientifico ( ex officio ) della
istituzione sede della sperimentazione nei casi di cui allall’’art. 1, comma 2, un
dirigente appartenente all’ all’assessorato alla sanità
sanità regionale o delle province
autonome;
Un esperto in materia giuridica e assicurativa o un medico legale legale
Un esperto in bioetica
Un rappresentante del settore infermieristico
Un rappresentante del volontariato per l’l’assistenza e/o associazionismo di tutela
dei pazienti
Agenzia Italiana del Farmaco – Sperimentazione ClinicaDM 12 Maggio 2006
art. 4 Organizzazione
Art. 4 comma 4. Il Comitato etico rende
pubblicamente disponibili le modalità di
valutazione e di adozione dei pareri, tra
cui il quorum necessario per la loro
espressione, che comunque deve essere
di almeno la metà più uno dei
componenti; le decisioni sono assunte
dalla maggioranza dei presenti aventi
diritto al voto.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaDichiarazione di Helsinki
E’ dovere del medico promuovere e salvaguardare la salute delle persone
Il progresso medico è fondato sulla ricerca la quale a sua volta si deve
basare in qualche misura su una sperimentazione che coinvolga soggetti
umani.
Lo scopo primario della ricerca medica che coinvolga soggetti umani è
quello di migliorare le procedure preventive, diagnostiche e terapeutiche e
di comprendere l’eziologia e la patogenesi della malattia.
Le considerazioni correlate con il benessere del soggetto umano
devono avere la precedenza sugli interessi della scienza e della società.
Agenzia Italiana del Farmaco – Sperimentazione ClinicaDirettiva 2001/83/CE del Parlamento Europeo e del Consiglio
del 6 Novembre 2001
Lo scopo principale delle norme relative alla
produzione, alla distribuzione e all’uso di medicinali
deve essere quello di assicurare la tutela della
sanità pubblica
(considerando 2)
Agenzia Italiana del Farmaco – Sperimentazione ClinicaIl farmaco rappresenta un bene per la
comunità e uno strumento privilegiato di tutela
della salute pubblica
Impegnarsi per migliorare la qualità
dell’assistenza farmaceutica significa tutelare
la Salute
considerata “ come diritto fondamentale
dell’uomo e interesse della collettività”
Art.32 Costituzione Italiana
Agenzia Italiana del Farmaco – Sperimentazione ClinicaGrazie per l’attenzione!
mf.barchetti@aifa.gov.it
Agenzia Italiana del Farmaco – Sperimentazione ClinicaPuoi anche leggere

















































