Raffaele Manna Verso una Cultura della Salute che accolga i pazienti con MR. L'esperienza del Policlinico Gemelli
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
XII GIORNATAMONDIALE MALATTIE RARE
22-23 febbraio 2019
Matera
Verso una Cultura della Salute che accolga i pazienti con MR.
L’esperienza del Policlinico Gemelli
Raffaele Manna
UOC Medicina Interna Columbus
Centro di ricerca delle febbri autoinfiammatorie e malattie rare
Fondazione Policlinico Gemelli IRCCS
ambulatorio.febbriperiodiche@policlinicogemelli.it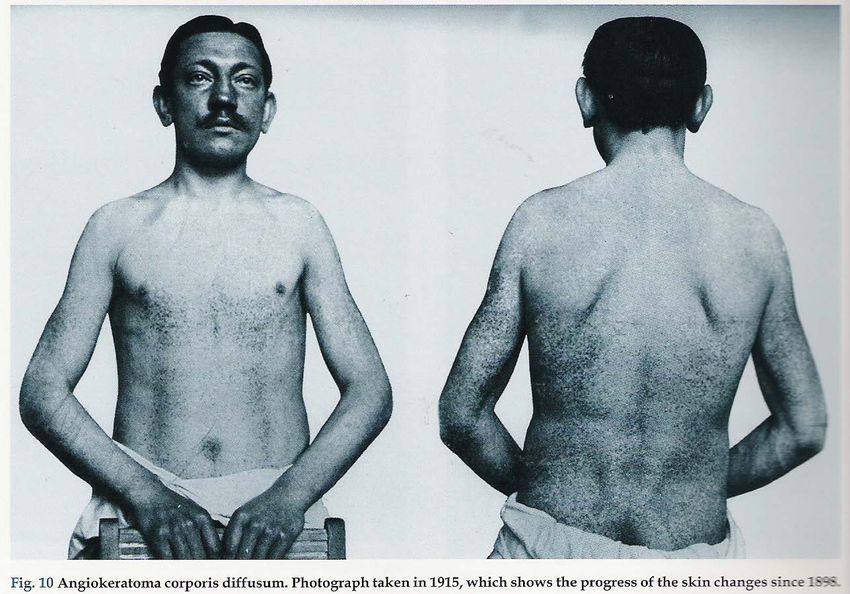
Quando senti rumori di zoccoli pensi ai cavalli? o alle zebre? Manifestazioni rare di malattie comuni sono più probabili che malattie rare Il problema è che le malattie rare possono essere “impostori” ORPHANET : LE MALATTIE RARE SONO QUELLE CHE NON SI CONOSCONO
Le malattie rare • colpiscono un piccolo numero di persone rispetto alla popolazione generale e questioni specifiche emargono in relazione alla loro rarità. • In Europa, una malattia è considerata rara quando colpisce 1 persona / 2000. Una malattia può essere rara in una regione, ma comune in un'altra. Ci sono > 6000 malattie rare e nuove malattie sono regolarmente descritte nella letteratura medica • Mentre l’80 % delle malattie rare sono malattie genetiche, ma non tutte le malattie rare sono malattie genetiche. • Esistono anche forme molto rare di malattie infettive, come malattie autoimmuni e tumori rari. • Ad oggi, la causa di molte malattie rare rimane sconosciute; molte malattie restano indefinite e senza una diagnosi.

La prima febbre periodica ereditaria 5 cases of recurrent "parossisms with severe abdominal pain with fever as high as 105 °F " all starting early in life The peritoneal signs were so severe that "emergency operation [had] been repeatedly urged." Pleuritic chest pain often accompanied the attacks. At least one of the patients had "urticarial" lesions near the ankle, and another had intermittent joint pains, usually monoarticular .

Centro delle Febbri Periodiche e Malattie Rare (1997) • Obiettivi: diagnosi e trattamento di malattie febbrili periodiche rare • Attività clinica: dignosi di pazienti con febbri periodiche di origine sconosciuta, provenienti da tutta Italia, e anche da Stati di Stati (Malta, Svizzera, Tunisia, Egitto) • Centro di riferimento per le febbri periodiche autoinfiammatorie • Attività di ricerca • De Benedetti F et al. N Engl J Med 2018;378:1908-1919 • Verrecchia Manna European Journal of Internal Medicine xxx (2016) xxx–xxx • Attività formativa: • Congresso Mondiale sulla FMF e SIAD, Roma 2008 • Master formativi annuali (prossimo giugno 2018) • Il CFP collabora con il Centro Amiloidosi di Londra (P. Hahwkins) Pavia (Prof Merlini) e partecipa alla rete europea di eccellenza di PRINTO e EUROFEVER (Gaslini).

Gene MeFV (Mediterranean FeVer) -16p13.3
1 2 3 4 5 6 7 8 9 10
Identificato col “positional cloning” (1, 2)
Il MeFV codifica per una proteina (95KD, 785aa) denominata marenostrina (da
Mare Nostrum cioè Mar Mediterraneo) o pyrina, espressa principalmente nei
neutrofili, monociti ed è implicata nell’inflammasoma, la linea di frontiera
delle difese immunitarie.
bZIP B-Box
NH2 PYD Coiled-coil B30.2 COOH
1) The French FMF consortium. 2) The international FMF consortium
Nature Genetics 1997; 17:25-32 Cell 1997; 90(4):797 807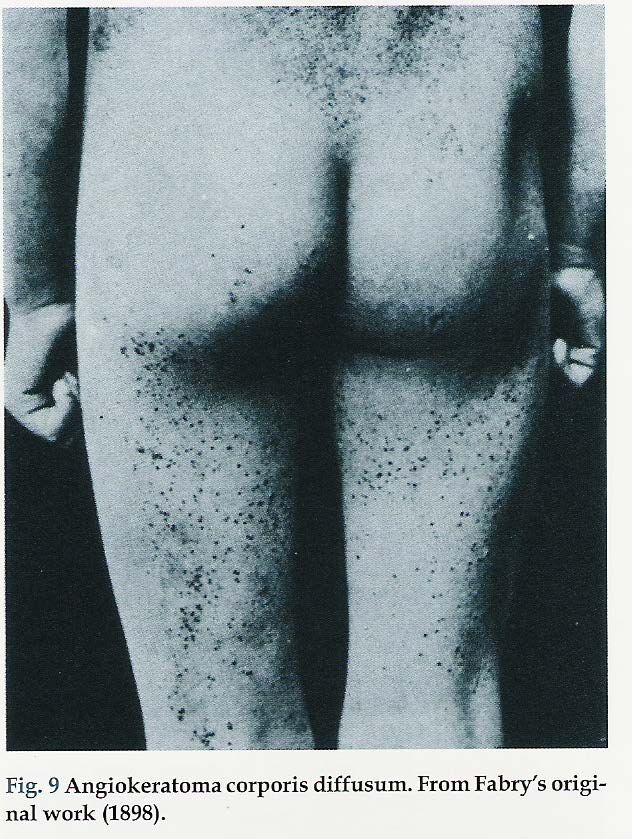
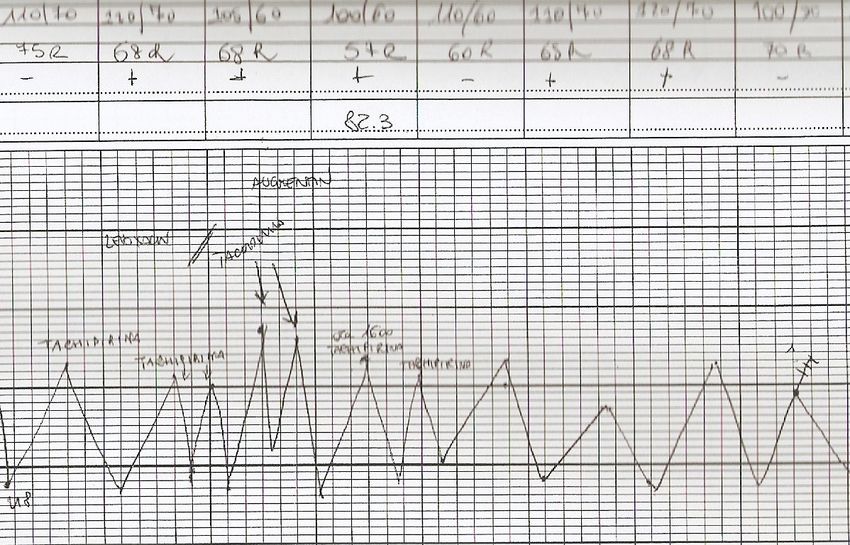
Curve termiche
nelle FEBBRI PERIODICHE EREDITARIE
R Manna 2010
M. La Regina, I. Touitou, G.
Gasbarrini, R. Manna
Sin dal 1998, >1300
pazienti con Febbre
ricorrente di origine
sconosciuta (FUO) sono
giunti al Centro di Ricerca
delle Febbri Periodiche” di
Roma.
Tra questi pazienti, fino al
2015, noi abbiamo
selezionato 321 pazienti
con FMF.
Il tipo di mutazioni MeFV e l'origine geografica dei pazienti, con una progressione
Nord-Sud, sembra rappresentare le ondate migratorie verso l'Italia nel corso dei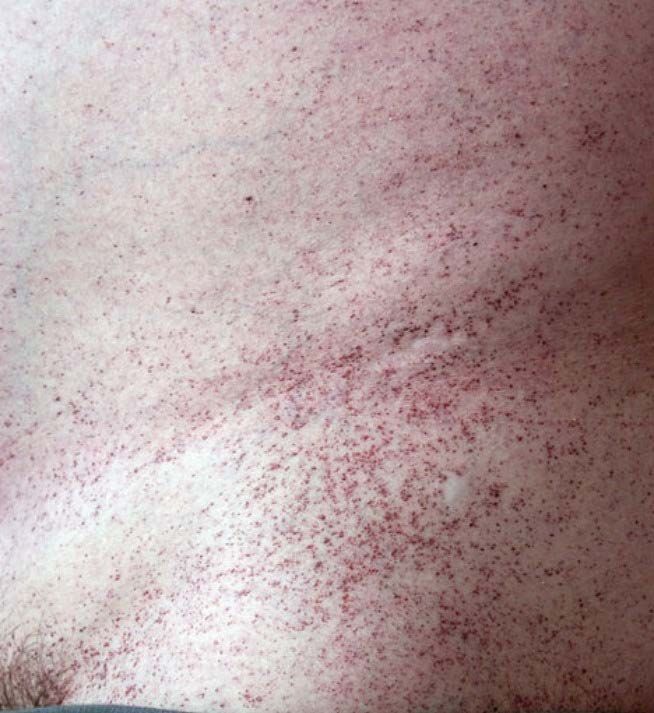
MANIFESTAZIONI Pazienti Manifestazioni cliniche
CLINICHE %
Febbre 93,75
Dolore Addominale 83,33
Dolore Toracico 40,27
Dolore Articolare 68,05
Lesioni cutanee 29,86
Aftosi 28,82
Mialgie 37,5
Interessamento Renale 13,54
Orchite 2,77
Solo il 6,25% hanno attacchi dolorosi senza febbre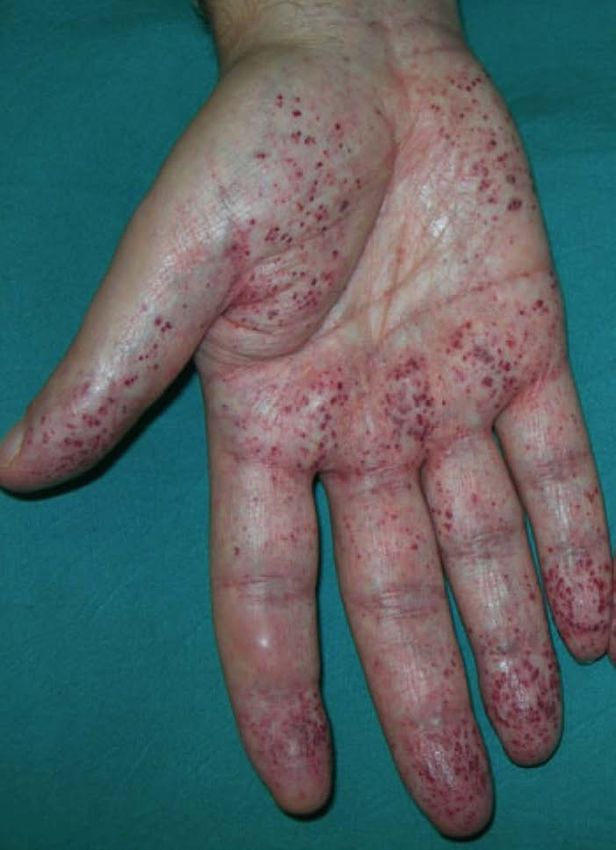
• Condizioni febbrili ricorrenti caratterizzate da episodi acuti di febbre con
brividi, in assenza di infezione
• FREQUENZA VARIABILE degli EPISODI: 2-3 attacchi / anno a 4 attacchi /
mese
• Gli episodi sono auto-limitati e con RECUPERO COMPLETO entro 3 - 30
giorni
• PERIODO INTERCRITICO: il paziente appare in buona salute, con normali
marcatori infiammatori; normale crescita ed energia di recupero nei
pazienti pediatrici
2001Perché dovremmo studiare Malattie autoinflammatorie? 1. Disturbi caratterizzati da episodi di infiammazione apparentemente non provocata senza autoanticorpi o linfociti antigene-specifici (McDermott & Kastner, Cell 1999) Febbri periodiche ereditarie 2. Mutazioni dei geni dell'immunità innata: • NLR o proteine dell'infiammasoma (La linea di frontiera della risposta immunitaria) • Proteine coinvolte nella regolazione della citochine. • Recettori delle citochine (D.I.R.A.) 3. Diversi fattori ambientali svolgono un ruolo di trigger come freddo, stress, cristalli o patogeni / pattern molecolari (PAMPS o DAMPS) 4. Manifestazioni cliniche diversificate
Immunità Innata vs Adattiva
• Velocissima: 4-96 ore, infiammazione • Risposta Specifica: consente a milioni di
immediata (febbre, sieropositività, ecc.) combinazioni di recettori Ag di rilevare un numero
• Risposta Aspecifica infinito di agenti
• Codificato da un gran numero di geni • Risposta Lenta: si sviluppa 4-15 giorni dopo
che codificano per recettori, linfochine, l'innesco:
complementi ecc. • Codificati da geni MHC, Immunoglobuline e T
linfociti
• Conservato nell'evoluzione sin dai
metazoi agli organismi multicellulari. • Proprio dei vertebratri, evoluto in 500 milioni di
anniFEBBRI PERIODICHE AUTOINFIAMMATORIE
Diseases Gene Protein Transmission
Chromosome
Familial Mediterranean Fever MEVF Pyrin AR
16p13.3
Periodic/recurrent Mevalonate Kinase Deficency MVK Mevalonate kinase AR
Fevers 12q24
TRAPS TNFRSF1A p55 TNF receptor AD
12p13
FCAS, MWS, CINCA CIAS1/NLRP3 Cryopyrin AD
NLRPs-related diseases 1q44
NLRP12-related periodic NLRP12 NLPR12 AD
fever 14p35
Granulomatous Blau’s syndrome CARD15/NOD2 CARD15 AD
disorders 16q12
PAPA syndrome PSTPIP1 15q24- PSTPIP1 AD
q25.1
Pyogenic disorders Majeed’s syndrome LPIN2 18p LPIN2 AR
CRMO (murine) PSTPIP2 18p PSTPIP2 AR
DIRA IL1RN 2p22 IL1Ra ADSpettro in espansione delle Malattie autoinfiammatorie
• Febbre Mediterranea Familiare
• TRAPS, MVK, CAPS (FCAS, Muckle Wells, CINCA)
• FEBBRE NAPL12 relata (FCAS2 ) Disordini piogenici
• PFAPA (esordio pediatrico e età adulta) PAPA, Majeed sdr, DIRA sdr :
• Artrite da cristalli (acido Deficit di antagonista del
urico, pirofosfati, asbesto, silice, colesterolo) recettore dell’ IL-1β
• Pericardite Ricorrente idiopatica
Disordini granulomatosi:
• Sindrome Schnitzler
Sindrome Blau e Early Onset
• CRMO (Chronic recurrent Multifocal
osteomyelitis) Sarcoidosis
• Morbo di Still dell’adulto (AOSD) CANDLE syndrome o Nakajo-
• Idrosadenite suppurativa Nishimura syndrome
• Morbo di Behçet (multifattoriale)
• Sindrome SAPHO
• Sindrome SWEET
R. MannaLa terapia profilattica della FMF
Fino al 1972 la terapia della FMF si basava sulla somministrazione
di sintomatici
S. Goldfinger.
Colchicine for familial Mediterranean fever.
NEJM 1972; 287(25):1302
Alcaloide contenuto nei semi del colchicum
autumnale, usato da secoli nel trattamento della gotta.
Riduce la chemiotassi e l’attivazione neutrofilica
Previene l’amiloidosi
Intolleranza: Diarrea (intolleranza al lattosio)
Effetti collaterali: aumento delle transaminasi etc
vera resistenza 5-10 % Το δακτυλον του Ερµητου
Ristretto indice terapeutico:
La giusta dose è ciò che differenzia un rimedio da un veleno.
ParacelsoCondizioni favorenti la tossicità della colchicina
In condizioni morbose concomitanti
Gastrite atrofica Leucopiastrinopenia da deficit di B12
Mielodisplasia Leucopiastrinopenia severa
Dieta vegana Leucopiastrinopenia da deficit di B12
Durante chemioterapie per linfomi Interazioni con le chemioterapie
Trapiantati di rene interazioni con CyA o altri ciclofillina inibitori
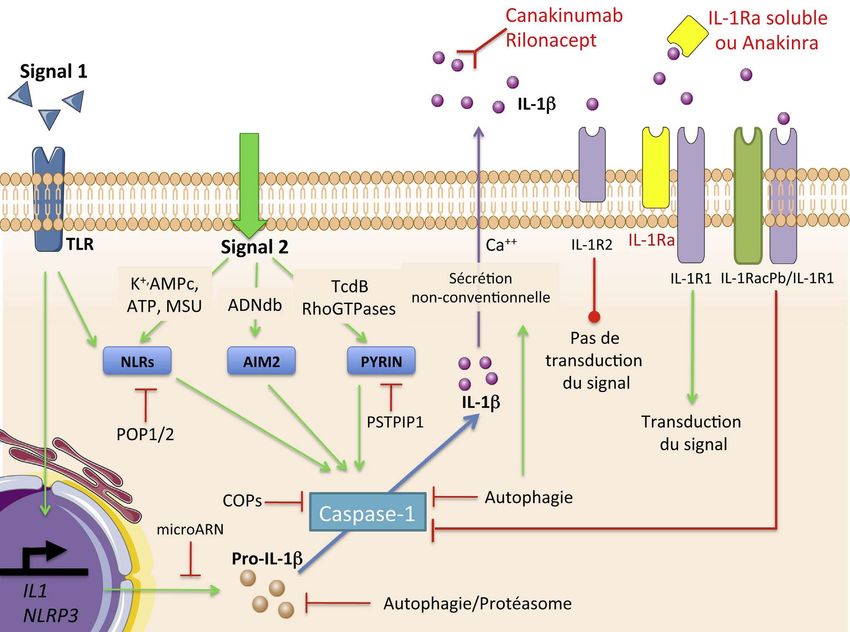
Depressione endogena primaria : Rischio suicidario (farmaco non dializzabile!)Current available biological agents targeting on IL-1β
378(20):1908-1919 May 17, 2018 Alla 16 settimana, i pazienti trattati con Canakinumab hanno avuto una risposta completa rispetto a quelli trattati con placebo: 61% vs 6% dei pazienti con FMF resistente alla colchicina (P
Filippo 34 anni Ott 2013 Riferito al nostro centro di Malattie Rare per la febbre ricorrente di origine sconosciuta (FUO) Trattamenti antibiotici sempre inefficaci Iniziata all’età di 5 anni, in maniera ricorrente con durata variabile da pochi giorni a più di un mese, accompagnata da dolori addominali talora diarrea Marzo / Maggio 2013: febbre intermittente con brivido (picco di 38,3 °C) iniziato dopo un episodio di diarrea e trattata con basse dosi di cortisone * Il paziente riferiva di aver una febbre prolungata ogni anno dall’età di 5 anni , spesso dopo una virosi acuta
Anamnesi patologica prossima (I)
Ricorrenza della febbre dopo la sospensione della terapia
steroidea Luglio 2013: ricoverato nell'Ospedale locale*
per FUO e sottoposto a una valutazione esaustiva
Esami normali : emocromo, elettroliti, funzione epatica, renale e
tiroidea, elettroforesi proteica, marcatori infiammatori (CRP, ESR, B2-
microglobulina, fibrinogeno, ferritina)
Autoimmunità (RF, c-ANCA, pANCA, ANA, anti-ENA, componenti del
complemento, anti-tireoglobulina, anti-TPO)
Patologie ematologiche e tumori maligni (negativa la biopsia del midollo
osseo, negativa la TAC di testa, collo torace-addome, eco per prostata e testicoli)
Sierologia per malattie infettive :
CMV, EBV, HIV, Toxoplasma, adenovirus, enterovirus, Widal-Wright e Weil
Felix, emocolture
Ecocardiogramma per i segni di endocardite
* Esami eseguiti nell’Ospedale localeOttobre 2013 - CFP: Approfondimento anamnestico
2007 Episodio di perdita di udito e acufene
nell'orecchio destro (2007) *
ipoacusia percettiva bilaterale per le alte
frequenze
2012 Episodio di vertigine oggettiva associata con
acufene persistente nell'orecchio sinistro
Il dermatologo esclude lesioni significative
?
Acroparestesie distali and andamento centripeto
*RMN cranica con evidenza di piccola cisti leptomeningea temporale destraDiagnosi dopo 29 anni
Ottobre 2013: test mirati per
1) per febbri auto-infiammatorie
(MEFV, TRAPS, MVK)
2) saggio enzimatico dell’attività α-galattosidasica
α-galattosidasi = 4.46 ± 0.21 (v.n. 66-88)
5% rispetto ai controlli
Analisi molecolare per confermare la diagnosi FD
Mutazione c907A>T plle303Phe (non ancora descritta)DIAGNOSIS
Age 17 34 11 13 32 15 20 58 16 32 31 16
33
Diagnostic 9 29 5 7 25 10 14 48 5 12 20 6
16
Delay
Hospitalization Y Y N N Y Y N N Y Y N Y
7
12/58 patients (20.7%) had fever/hyperthermia
Fever
Onset Age 8 5 6 6 7 5 6 10 10 10 6 10
7.4
Age 18 36 10 10 34 20 19 16 30 30 30n 18
disapparance
Flares /month 1-3Algoritmo per FUO e RFUO
Malattia di Anderson-Fabry 1898: W. Anderson descrisse un paziente di 39 anni che aveva un’eruzione purpurica sul tronco, sullo scroto ed agli arti. La malattia era comparsa in età infantile e si erano sviluppate vene varicose, rettorragia e albuminuria. Anderson chiamò questa condizione "angiokeratoma” e suggerì che vi potesse essere un’alterazione generalizzata del sistema vascolare 1898 J. Fabry descrisse la stessa patologia di un ragazzo di 13 a, denominandola "purpura haemorrhagica nodularis". Fabry mantenne il suo interesse per il paziente e pubblicò i risultati dell'autopsia dopo la morte del paziente nel 1930.
1930 Il paziente muore all’età di 43 anni, per una non meglio definita affezione “polmonare”. J. Fabry descrive i reperti autoptici: ipertrofia cardiaca, edema polmonare, reni piccoli e muore dopo qualche mese
Manifestazioni dermatologiche: ANGIOCHERATOMI
CONFRONTO DEGLI ASPETTI CLINICI DELLA FEBBRE MEDITERRANEA FAMILIARE E DELLA MALATTIA DI FABRY. ASPETTI CLINICI FEBBRE MEDITERRANEA FAMILIARE MALATTIA DI FABRY ETA’ ALL’ESORDIO
Malattia di Fabry: definizione e frequenza
Malattia da Deficit di α-galattosidasi A con accumulo
lisosomiale di globotriaosilceramide (Gb3)
Malattia ereditaria, legata al cromosoma X
l’incidenza stimata 1:40000-117.000
prevalenza nello screening neonatale 1:4000
Interessamento sistemico e/o di un unico apparato
Decorso lentamente progressivo
L’aspettativa di vita dei pazienti maschi è 58.2 anni, vs 74.7
nella popolazione generale USA*.
L’aspettativa di vita delle femmine è 75.4 anni vs 80 nella
popolazione generale USA*
*Fabry RegistryLa Malattia di Fabry ‘Il Grande Impostore”
*French Observatoire in Internal Medicine Departments
O. Lidove Clin Genet 2012: 81: 571–577DIAGNOSI • La diagnosi viene complicata dalla rarità delle malattia, dal numero degli organi coinvolti e dai sintomi non specifici • Il paziente tipico viene visitato da circa 10 specialisti, nel corso di 10 anni, prima che venga correttamente diagnosticata la malattia • Ritardo medio alla diagnosi : 13 anni • Nel bambino viene spesso posta la diagnosi di “dolori di crescita” o di connettivite, artrite idiopatica giovanile “Febbri di crescita” “dolori addominali” sine materia
Malattia di Anderson Fabry
• Malattia rara
• Prevalenza di 1:117,000 maschi
Branton MH, 2002; Meikle PJ, 1999; Houge G, 2005
Deutsches Ärzteblatt International⏐Dtsch Arztebl Int 2009; 106(26): 440–7La Trasmissione della Malattia dal padre
•Il maschio affetto dalla
Malattia di Anderson-Fabry
trasmette il gene a tutte le
figlie femmine ma a
nessuno dei figli maschi
La femmina eterozigote
manifesta diversi gradi di
coinvolgimento patologico che
vanno dalla scarsa alla piena
espressione della malattia2La Trasmissione della Malattia dalla madre
•Le femmine eterozigoti
hanno il 50% di possibilità
ad ogni gravidanza di
trasmettere il gene ai propri
figli maschi e femmine.
•Il maschio riceve il gene
mutato unicamente dalla
madre
•Il maschio presenta
sempre la malattiaEvoluzione naturale della malattia di Fabry
Manifestazioni di malattia
Insufficienza
d’organo
Depositi tissutali
Incremento di GB3
Andamento temporale
0-15 anni 15-30 anni > 30 anni
- Angiocheratoma - Acroparestesie
- Acroparestesie - sintomi gastrointestinali - Insufficienza renale
- Sintomi Gastrointestinali
- Sintomi cardiaci
Intolleranza a caldo - Sintomi renali
- proteinuria -
Ipo/iperidrosi - Ipo/iperidrosi Ictus
- Cornea verticillata,Algoritmo diagnostico per M di Fabry
SINTOMI PRECOCI QUADRI rilevanti
• Acroparestesie • Proteinuria, insuff. Renale
• Dolore addominale • Ipoacusia inspiegata
• Febbre/ipertermia • Cardiomiopatia ipertrofica
• Intolleranza al caldo • TIA o STROKE
Markers suggestivi
si • Cornea verticillata si
• Angiocheratoma
• Storia familiare di Malattia di
Fabry
SI
MASCHI FEMMINE
Analisi enzimatica dell’α galattosidasi + Mutazioni del gene Gal
Conferma lysoGB3, istologia, analisi in silico e in vitroTerapia enzimatica sostitutiva (ERT)
Disponibile dal 2001: enzima ricombinante
Due farmaci:
1) agalsidasi alfa (REPLAGAL)
0,2 mg/kg ev in 40’ ogni 14 gg
prodotto da fibroblasti umani in cultura
2) agalsidasi beta (FABRAZYME)
1 mg/kg ev in 2-4 h ogni 14 gg
prodotto da cellule ovariche Hamster
• Diversa glicosilazione e diversa contenuto di acido sialico e mannosio
• Le differenze in termini di efficacia tra i due farmaci sono molto dibattute1
1 Vedder AC, Linthorst GE, Houge G, et al. Treatment of Fabry disease: Outcome of a comparative trial with agalsidase alfa or beta at a
dose of 0.2 mg/kg. PLoS One. 2007;2(7):e598.Malattia di Fabry :terapia orale chaperonica Trapia orale chaperonica Migalastat Per le mutazioni amenable (50%) -Approvato dall’ EMEA e dallAIFA
N. Engl. J. Medicine aug 2016 : Migalastat GALAFOLD ®
Original Article
Treatment of Fabry’s Disease with the
Pharmacologic Chaperone Migalastat
Dominique P. Germain, M.D., Ph.D., Derralynn A. Hughes, M.D., Kathleen
Nicholls, M.D., Daniel G. Bichet, M.D., Roberto Giugliani, M.D., William R.
Wilcox, M.D., Ph.D., Claudio Feliciani, M.D., Suma P. Shankar, M.D., Ph.D., Fatih
Ezgu, M.D., Hernan Amartino, M.D., Drago Bratkovic, M.D., Ulla Feldt-
Rasmussen, M.D., Khan Nedd, M.D., Usama Sharaf El Din, M.D., Charles M.
Lourenco, M.D., Maryam Banikazemi, M.D., Joel Charrow, M.D., Majed
Dasouki, M.D., David Finegold, M.D., Pilar Giraldo, M.D., Ozlem Goker-Alpan, M.D.,
Nicola Longo, M.D., C. Ronald Scott, M.D., Roser Torra, M.D., Ph.D., Ahmad
Tuffaha, M.D., Ana Jovanovic, M.D., Stephen Waldek, M.D., Seymour
Packman, M.D., Elizabeth Ludington, Ph.D., Christopher Viereck, Ph.D., John
Kirk, D.Sc., Julie Yu, Ph.D., Elfrida R. Benjamin,
N Engl J Med Ph.D., Franklin Johnson, M.Sc.,
Volume 375(6):545-555
David J. Lockhart, Ph.D., Nina Skuban, M.D., Jeff Castelli, Ph.D., Jay Barth, M.D.,
August 11, 2016
Carrolee Barlow, M.D., Ph.D., and Raphael Schiffmann, M.D.CONCLUSIONI e
PROSPETTIVE-1
• Necessario ridurre i ritardi diagnostici delle FUO ricorrenti
implementando negli algoritmi diagnostici anche marcatori moderni ad
elevata specificità come test genetici, ed enzimatici, come
alfagalattosidasi etc).
• Per la Malattia di Fabry, la diagnosi precoce è importante perché ora ci
sono farmaci biologici che possono stabilizzare o invertire il decorso di
Malattia
• Malattie rare sono presenti nel gruppo con FUO ricorrenti (malattie
autoinfiammatorie, malattie metaboliche genetiche) E’ nato un
progetto per la diagnosi delle FUO ricorrenti, per la diagnosi di
malattie rare ( M di Fabry etc)….CONCLUSIONI e PROSPETTIVE-2 1. Nelle malattie autoinfiammatorie monogeniche il percorso comune finale è il rilascio di IL-1b e di altre citochine infiammatorie (IL-6) da essa indotte. 2. I nuovi farmaci biologi anti- IL-1b (Anakinra e Canakinumab) sono potentemente efficaci nelle condizioni autoinfiammatorie monogeniche e anche in molte forme acquisite. 3. L’antagonista recettoriale dell’IL1b (Anakinra) è efficace nelle pericarditi ricorrenti idiopatiche, resistenti alla colchicina e nella sdr di Schnitzler. 4. Il monoclonale umano ad alta affinità anti IL1b (Canakinumab) è efficace nel controllare le febbri periodiche ereditarie, il Morbo di Still e riduce le complicazioni dell’aterosclerosi precipitate dall’infiammazione.
Riconosce solo quello che si conosce
La nostra forza è il….. Gruppo •Prof D. Antuzzi Enzimatica •Prof. G. Gambaro Nefrologia •Prof A. Gasbarrini Gastroenterologia •Prof M. Genuardi Genetica •Prof. A. Morrone Genetica •Dr M.G. Pomponi Genetica •Dr. S. Conti ORL •Dr. R. Flore Angiologia •Dr. R. Fasciani Oculistica •Dr. F. Graziani Cardiologia •Dr. R. Marano RMN cardiaca •Dr. A. Paolucci Infusioni •Dr. G. Pasciuto Pneumologia •Dr L. Sicignano Logistica •Dr. G. Silvestri Neurologia •Dr. E. Verrecchia Coordinazione Trial e Registri
GRAZIE dell’attenzione
Maurizio Genuardi
Giovanni Neri
Fiorella Gurrieri
M.Grazia Pomponi
Roberta Pietrobono
Donato Rigante
Giovanni Ghirlanda
Centro di Ricerca delle Febbri periodiche
Istituto di Medicina Interna * L. Luca Sicignano
Istituto di Pediatria Elena Verrecchia
Istituto di Medicina Genomica *
Fondazione P. Gemelli IRCS
Università CattolicaThanks
FMF: Difficoltà della diagnosi
• Non risposta alla colchicina
• 1 mutazione o nessuna
mutazione vasculiti
BD
• Associazione con altra
malattia FMF JIA
• Vasculiti (H-SP, PAN) MS
• IBD IBD
• AS: Spondilartriti AS
• BD: Behçet’s Disease
• MD Majeed SyndromePattern delle Febbri autoinfiammatorie ereditarie
R Manna 2010La terapia profilattica della FMF
Fino al 1972 la terapia della FMF si basava sulla somministrazione
di sintomatici
S. Goldfinger.
Colchicine for familial Mediterranean fever.
NEJM 1972; 287(25):1302
Alcaloide contenuto nei semi del colchicum
autumnale, usato da secoli nel trattamento della gotta.
Riduce la chemiotassi e l’attivazione neutrofilica
Intolleranza: Diarrea (intolleranza al lattosio)
Effetti collaterali: aumento delle transaminasi etc
vera resistenza 5-10 % Το δακτυλον του Ερµητου
Ristretto indice terapeutico:
La giusta dose è ciò che differenzia un rimedio da un veleno.
ParacelsoFARMACOCINETICA DELLA COLCHICINA
La biodisponibilità della colchicina può mostrare ampie differenze individuali dal
24% all'88% (media 45%) in volontari sani.
La colchicina raggiunge il picco di concentrazione plasmatica circa 1 ora dopo la
somministrazione di una singola dose orale, mentre il suo accumulo all'interno
delle cellule, come granulociti o leucociti mononucleati, può richiedere 2 giorni.
L’emivita è 9 giorni.
Lesioni della mucosa digiunale (sito principale di assorbimento) sono state
associate all'uso di colchicina a lungo termine (deficit di B12/vit D)
Deficit di assorbimento conseguente a diarrea infrenabile
Cerquaglia C, and Manna R. Pharmacological and clinical basis of treatment of FMF.
Curr Drug Targets Inflamm Allergy. 2005;4(1):117.OBIETTIVI DELLA TERAPIA
Esempio di profilo temporale 1) Prevenire gli attacchi;
degli attacchi di febbre durante 2) Controllare l’infiammazione subclinica nei
l'anno (a) e del singolo attacco
febbrile nelle ore (b) periodi intercritici.
3) Prevenire l’insorgenza dell’amiloidosi•Depolimerizzatore reversibile dei microtubuli, impedisce la migrazione e l’attivazione dei neutrofili •Trasportata nel compartimento citoplasmatico dei linfomononucleati; i livelli circolanti non sono utili •la colchicina è il farmaco di prima scelta In grado di prevenire l’attacco di FMF •L’assunzione continuativa di colchicina è in grado di prevenire lo sviluppo di amiloidosi; e l’insorgenza di insufficienza renale; • Ricomparsa dell’attacco febbrile in caso di sospensione del farmaco.
Precauzioni di impiego •Insufficienza epatica severa: rischio di accumulo del farmaco • Insufficienza renale cronica: per la diminuita eliminazione • Età avanzata: rischio maggiore di tossicità • Miopatie preesistenti (Duchenne, iperCPK): rischio aggravamento della malattia • Colestasi: rischio di accumulo •In tutti questi casi la colchicina è stata somministrata ma con riduzione della dose e attenta osservazione e monitoraggio clinico; • GRAVIDANZA: nessuna documentazione di rischi aumentati; il 15 % riduce la dose, qualche paz si autosospende la terapia. L’amniocentesi è stata consigliata; l’aborto non è giustificato.
Interazioni farmacologiche
Con i farmaci che inibiscono P450-3A4 / P-Gp
Farmaci che aumentano la tossicità della colchicina
• Macrolidi: claritromicina (azitromicina consentita)
• Ciclosporina: inibitore della calcineurina
• Azolici: come Ketoconazolo o itraconazolo: (terbinafina consentita)
• Statine: (solo la fluvastatina consentita)
• Ritonavir
• Omeprazolo inibitori pompa protonica (pantoprazolo consentito)
• Diltiazem: antipertensivo calcio-antagonista
• Gestodene: lieve incremento dei livelli di colchicina
• Succo di pompelmo, melograno: inibisce il P- glicoproteina aumentando
l’assorbimento e la tossicità
Farmaci che interagiscono con la colchicina in modo imprevedibile
•Warfarin: possono aumentare i livelli di warfarin o quelli della colchicina
•Amiodarone: antiaritmico di classe VPosologia ed efficacia
Dose: fino a 0,03 mg/Kgpc/die
Emivita: 9 giorni
Range terapeutico: 2-3 settimane
1) Pz RESPONDERS:
RISPOSTA COMPLETA: scomparsa completa dei sintomi: >60 %
RISPOSTA PARZIALE: riduzione della frequenza, dell’intensità e della durata degli
attacchi. (30-35%)
2) Pz NON Responders (5-10%): persistenza di attacchi >1 volta ogni 3 mesi (Lidar),
nonostante il trattamento regolare con colchicina a 3 (2) mg di al giorno ( o 2 mg nei
bambini); (11% compliance irregolare).
LA COLCHICINA, in linea generale, è SICURA e BEN TOLLERATA, tuttavia:
5-30 % DEI PAZIENTI è INTOLLERANTE ALLA COLCHICINA;FACTORS AFFECTING THE COLCHICINE RESPONSE
Intolleranza e tossicità alla colchicina
Effetti collaterali principali: non sempre legata ad alti livelli ematici
• Gonfiore addominale
• Diarrea
• Vomito
•Aumento delle transaminasi
•Aumento degli enzimi muscolari
•Anemia o leucopenia (deficit di vit B12)
•Azospermia (5%-, reversibile)
•Alopecia (1/1000)
•Tossicità del Sistema nervoso centrale o periferico (vertigini, cefalea)
•Danno multiorgano fino alla CID (in caso di sovradosaggio erroneo o suicidario)
(Dialisi inefficace; utili i frammenti FAB anticolchicina)Condizioni favorenti la tossicità della colchicina
In condizioni morbose concomitanti
Gastrite atrofica Leucopiastrinopenia da deficit di B12
Mielodisplasia Leucopiastrinopenia severa
Dieta vegana Leucopiastrinopenia da deficit di B12
Durante chemioterapie per linfomi Interazioni con le chemioterapie
Trapiantati di rene interazioni con CyA o altri ciclofillina inibitori
Depressione endogena primaria : Rischio suicidario (farmaco non dializzabile!)Miglioramento della tolleranza gastrointestinale
I disturbi gastrointestinali possono essere dovuti a:
•Intolleranza al lattosio: la colchicina determina riduzione dei livelli di lattasi; la diarrea può
a sua volta ridurre l’assorbimento della colchicina Dieta senza o con poco lattosio
•Contaminazione batterica dell’intestino tenue: (normalmente sterile)
Terapia decontaminante con disinfettanti intestinali e probiotici *
•Malattia Celiaca: può coesistere indipendentemente dall’FMF, ma la colchicina può
svelare o peggiorare la celiachia Dieta aglutinata
•Gastrite da Helicobacter Piloryii: può coesistere indipendentemente dall’FMF, ma la
colchicina può svelare la gastrite da HP, che può ridurre l’efficacia della colchicina.
Eradicazione con triplice terapia antibiotica
•Gastrite erosiva: citoprotettivi (alcuni inibitori di pompa interferiscono col CYP34A)
* Verrecchia E. Manna RSmall Intestinal Bacterial Overgrowth Affects the Responsiveness to Colchicine in FMF Mediators of Inflammation 2017Follow-up ( dopo il 1°e 4° mese) Indici di infiammazione: VES, proteina C reattiva, fibrinogeno, SAA per la Prevenzione dell’amiloidosi: 1-4 volte l’anno Funzionalità renale: creatinina, azoto ureico, esame urine, microalbuminuria Emocromo con formula (e vit B12) Funzionalità epatica: AST, ALT, gamma-GT, fosfatasi alcalina, bilirubina Enzimi muscolari: CPK, LDH, troponina T
PRAS’ SEVERITY INDEX
Malattia severa >9
Malattia intermedia 6-8
Malattia moderata 3-5
Pras M, What is FMF, FMF International Conference,
2000 AntalyaResponse to colchicine treatment
according to the M694V mutation
Homozygotes Heterozygotes Others
Complete response 22 (36.1) 43 (54.4) 56 (70.0)
Incomplete response 28 (45.9) 33 (41.7) 19 (23.7)
Unresponsiveness*** 11 (18.0) 3 (3.9) 5 (6.3)
Mean colchicine dose, 1.42 ± 0.33 1.35 ± 0.48 1.31 ± 0.67
(Soylemezoglu et alJ Rheumatol. 2010 Jan;37(1):182-9• Definizione di resistenza alla colchicina su base clinica: se ha> 6 attacchi tipici FMF/anno o >3 tipici
attacchi FMF entro 4-6 mesi.
• In caso di attacchi incompleti con un aumento di almeno 2 dei 3 reagenti di fase acuta (CRP, ESR, SAA)
la risposta è considerata parziale e richiede trattamenti aggiuntivi o alternativi alla colchicina
• In caso di resistenza alla colchicina, gli inibitori IL-1β con emivita medio-lunga (es. Canakinumab)
devono essere considerati.
• Poiché la colchicina è l'unico farmaco comprovato contro l'amiloidosi secondaria, deve essere
proseguita il più a lungo possibile insieme al farmaco anti IL-1β
*“Hentgen V, et al. Semin Arthritis Rheum. 2013;43:387-91.Different approaches for assessing colchicine response
1
2
3
4
5
Manna 19 Maggio 2018Considerazioni sulla colchicino-
resistenza
• Alcuni pazienti non possono essere trattati con la dose di 2 mg / die Inoltre la definizione
non tiene conto della gravità degli attacchi.
• Il criterio FMF 50 definisce la risposta In caso riduzione >50%, ma non fornisce
informazioni sui sintomi rimanenti
• Ulteriori 6 criteri forniti da Ben Chetrit includono: 1. variazione della frequenza degli
attacchi, 2. variazione nella durata degli attacchi, 3. valutazione globale paziente /
genitore della gravità della malattia, 4. valutazione globale del medico della gravità della
malattia, 5. variazione negli attacchi di artrite e 6. variazione delle proteine della fase
acuta . La risposta è definita in caso di effetto su 5/6 criteri
• Se gli attacchi sono incompleti, è necessario inserire anche l’aumento di almeno 2 dei 3
reattanti di fase acuta per definire la resistenza
• Alcuni pazienti che assumono colchicina possono non tollerare attacchi rari o possono
avere un aumento del rischio di amiloidosi secondaria; quindi la terapia aggiuntiva o
alternativa può essere giustificata
Manna 19 Maggio 2018Alternative terapeutiche FANS Effetto di soppressione dell’infiammazione solo se somministrato nelle fasi precoci dell’attacco. Scarsa risposta nelle forme più severe. Come Antipiretici Per controllare le riaccensioni perimestruali della FMF. Steroidi nella fase acuta dell’attacco auto-infiammatorio, con ottima risposta nella PFAPA e nella FMF. Somministrabili per os o i.m. Abbreviano gli intervalli intercritici e, a lungo andare, favorire gli effetti collaterali (aumento di peso, IGT o diabete metasteroideo, ipertensione);
Take Home Message La colchicina è la terapia di prima linea nei pazienti affetti da FMF, è sicura ed efficace, anche in gravidanza; Il suo utilizzo può determinare effetti collaterali frequenti e controllabili, quali la diarrea, ma anche effetti collaterali più rari e reversibili, come l’azoospermia (eseguire spermiogramma prima dell’inizio della tp); La conoscenza della farmacologia della colchicina può evitare danni gravi legati ai livelli raggiunti per l’interazione con altri farmaci metabolizzati dal CYP3A4; Nei pazienti non responders valutare la compliance alla terapia del paziente ed escludere l’intolleranza al lattosio, la SIBO, l’Helicobacter Pyl. In caso di resistenza alla colchicina, il Canakinumab si è dimostrato efficace in aggiunta alla colchicina, a dose tollerabile. Gli anti IL1β sono l’unica risorsa disponibile in casi di totale intolleranza alla colchicina.
Puoi anche leggere

















































