Guida ai reparti per manipolazioni cellulari
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Guida ai reparti per manipolazioni cellulari
A P S A R A l Guida ai reparti per manipolazioni cellulari l Indice
Indice
03 Introduzione
04 Il concetto di contenimento
05 Il laboratorio a contenimento
07 La cabina di sicurezza biologica
07 Classe I
07 Classe II
08 Classe III
09 Il concetto di Cleanroom
10 Le Good Manufacturing Practice (GMP)
112 Il controllo ambientale della cleanroom
12 Monitoraggio dell’impianto di condizionamento
13 Monitoraggio particellare
13 Monitoraggio microbiologico
14 Una manipolazione, un ambiente
15 Quale ambiente scegliere
15 Esempi di layout
17 Glossario
19 Referenze Apsara
Assinggroup
23 AM Instruments Groupne clinica
www.assing-group.it
www.apsara.it
Edizione 02 | 01/2008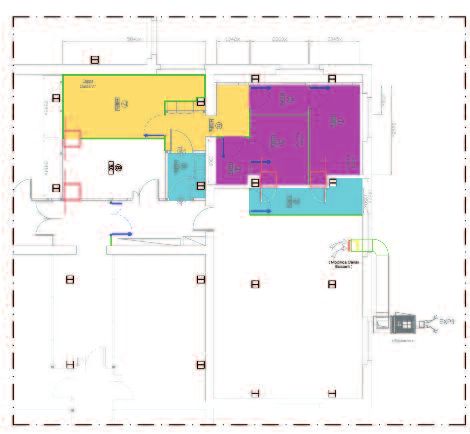
A P S A R A l Guida ai reparti per manipolazioni cellulari l Introduzione
Introduzione
Negli ultimi anni le manipolazioni ex vivo di cellule di origine animale o umana
da oggetto di ricerca accademica sono progressivamente diventate anche
oggetto di sperimentazione clinica. Il passaggio ha portato con sé la progressiva
introduzione di concetti tipicamente legati al settore farmaceutico tradizionale
(es. cGMP, validazione...) che si sono affiancati a quelli definiti dalle normative
di sicurezza (es. rischio biologico) che, negli stessi anni, si sono andate affermando.
L’utente ultimo dei locali dove le manipolazioni cellulari vengono effettuate,
ovvero lo scienziato ed il medico, può trovarsi disorientato davanti a questo
scenario. Questo fatto può portare a difficoltà di comprensione, e a volte
a fraintendimenti, con coloro che sono chiamati a progettare e a costruire
laboratori per attività di biologia cellulare. La corretta progettazione dei locali
costituisce infatti la base dove l’attività di sperimentazione clinica
può svilupparsi garantendo la sicurezza dell’operatore, dell’ambiente
e del paziente a cui sono destinate le cellule manipolate.
Con riferimento alle leggi e alle direttive italiane ed internazionali, questa guida
affronta brevemente alcuni concetti fondamentali quali ad esempio il concetto
di contenimento biologico di cleanroom, nella nostra esperienza spesso confusi,
definendo in base all’attività svolta il tipo di ambiente di lavoro necessario.
Ci auguriamo che queste pagine siano utili agli scienziati e ai ricercatori
e che soprattutto servano di stimolo all’approfondimento di concetti ormai
indispensabili per passare dalla fase di ricerca a quella di sperimentazione clinica.
Apsara srl
l 03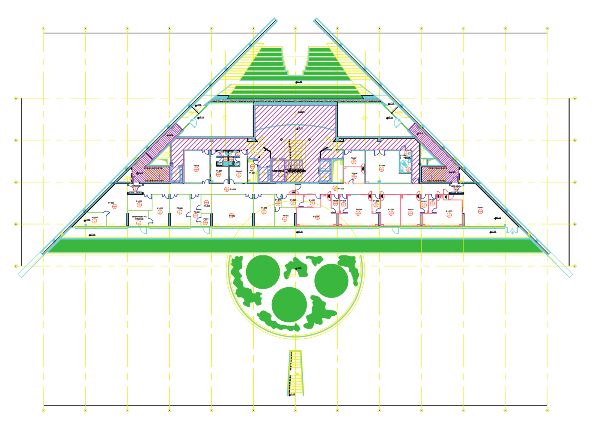
A P S A R A l Guida ai reparti per manipolazioni cellulari l Il concetto di contenimento
Il concetto di contenimento
Il termine “contenimento” è usato per descrivere l’insieme dei metodi che per-
mettono di manipolare o conservare materiale potenzialmente infetto. Scopo
del contenimento è quindi di ridurre o eliminare l’esposizione degli operatori e
dell’ambiente ad agenti pericolosi attraverso la costruzione di ambienti con
determinate caratteristiche, l’impiego di strumenti con particolari requisiti e
l’adozione di specifiche procedure di comportamento.
Figura 1
Concetto di
contenimento
Figura 2
Contenimento
primario e
secondario
Il cosiddetto contenimento primario, ovvero la protezione del personale e del
laboratorio dove avviene la manipolazione, è realizzato dall’uso di specifiche
procedure di manipolazione e dall’uso di strumentazione adatta. Il contenimento
secondario, ottenuto combinando il design dei laboratori e appropriate
procedure operative, ha invece lo scopo di proteggere l’ambiente all’esterno del
laboratorio di manipolazione.
La descrizione delle procedure operative (es. procedura smaltimento rifiuti) e dei
mezzi di protezione individuali (es. occhiali di sicurezza) richiesti o consigliati per
realizzare un appropriato contenimento non fanno parte dello scopo di questa
guida.
Vengono quindi considerate nei prossimi paragrafi solo le caratteristiche
costruttive dei laboratori e delle cabine di sicurezza biologica.
l 04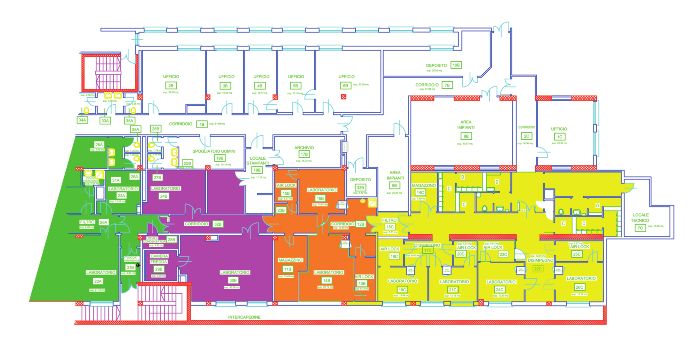
A P S A R A l Guida ai reparti per manipolazioni cellulari l Il concetto di contenimento
Il laboratorio a contenimento
In questi ultimi anni molto si è molto confinato di microrganismi genetica-
discusso riguardo alle caratteristiche mente modificati.
dei laboratori a di contenimento e Il decreto prevede che sia l’utilizzatore
varie sigle sono state utilizzate a a valutate il rischio del microrganismo
seconda delle normative italiane, geneticamente modificato e quindi
europee o americane di riferimento. scegliere il livello di contenimento
Sigla Norma/istituzione di riferimento Note
1, 2, 3, 4 D.Lgs. 626/94 patogeni “wild type”
1, 2, 3, 4 D.Lgs. 206/01 patogeni geneticamente modificati
PCL1, PCL2, PLC3, PCL4 UNI EN 12128, standard europeo
P1, P2, P3, P3 NIH (National Institute of Health, USA) superata
BL1, BL2, BL3, BL4 CDC (Center for Desease Control, USA) standard americano che sostituisce quello dell'NIH
Consideriamo innanzitutto la tra i 4 elencati. Tabella1
normativa italiana. Lo standard ISO EN 12128, dove Sigle e numerazione
laboratori di
Il documento di legge principale che l’acronimo PCL ha il significato di contenimento
parla di rischio biologico ed introduce Phisycal Containment Level, definisce
il concetto di contenimento è il D.Lgs anch’esso 4 livelli di contenimento.
626/94 che tratta il rischio biologico Tali livelli di contenimento rispecchiano
al Titolo VIII e suddivide nell’ allegato i 4 livelli indicati dal Center for
XI gli agenti biologici in 4 classi. Desease Control americano (ricordiamo
Conseguentemente l’allegato XII che la sigla BL corrisponde a
(specifiche sulle misure di conteni- Biosafety Level) e i 4 livelli indicati
mento e sui livelli di contenimento) e dalla ormai superata classificazione
l’allegato XIII (specifiche per processi del National Institute of Health
industriali) elenca le specifiche dei del 1999.
corrispondenti livelli di contenimento. Nella tabella seguente sono riassunti,
E’ importante notare che la classifica- per alcuni dei principali aspetti tecnici
zione degli agenti biologici considera e strutturali dei laboratori, i requisiti
gli agenti biologici “wild type” ovvero prescritti dal D.Lgs 626/94, dal D.Lgs
come esistenti in natura e non quelli 206/01 e dalla UNI EN 12128.
manipolati geneticamente. Di questi
ultimi si occupa il D.Lgs 206/01, che
specifica i criteri per l’impiego
l 05A P S A R A l Guida ai reparti per manipolazioni cellulari l Il concetto di contenimento
Filtrazione HEPA Livello di contenimento
1 2 3 4
D.Lgs 626/94 All. XII (1) no sì sull’aria emessa sì sull’ aria immessa
e sull’aria emessa.
D.Lgs 626/94 All. XIII (1) no facoltativo sì
D.Lgs 206/01 no no sì sull’aria emessa se la trasmissione sì sull’ aria immessa
del patogeno è per via aerea e sull’aria emessa.
UNI EN 12128 no no sì sull’aria emessa sì sull’ aria immessa
e sull’aria emessa doppio filtro.
Laboratorio in Livello di contenimento
depressione 1 2 3 4
D.Lgs 626/94 All. XII (1) no raccomandato sì
D.Lgs 626/94 All. XIII (1) no facoltativo sì
D.Lgs 206/01 no no sì, solo se la trasmissione sì
è per via aerea
UNI EN 12128 no no facoltativo sì
Presenza autocla- Livello di contenimento
ve 1 2 3 4
D.Lgs 626/94 All. XII (1) (2) (2) (2)
D.Lgs 626/94 All. XIII (1) (2) (2) (2)
D.Lgs 206/01 nel sito nell’edificio sul piano in laboratorio, passante
UNI EN 12128 no no sì, in zona in laboratorio, passante
Tabella 2 Confronto requisiti di contenimento leggi italiane e standard europei
Una categoria particolare di laboratori > sistema di condizionamento
a contenimento sono quelli destinati separato dall’impianto
alla preparazione di chemioterapici centralizzato (almeno 6 vol. di aria
e antiblastici che devono rispettare primaria/ora)
quanto previsto al Titolo II > pressione negativa rispetto
del Dlgs 626/94. ambienti esterni
Dato che tali laboratori > utilizzo di cappe a flusso laminare
esulano dagli scopi di questa guida, di classe II con lampada UV
si ricordano solamente le caratteristiche > necessità punto di decontamina
principali dei locali: zione del personale
(1) il D.Lgs. 626/94 non fornisce specifiche indicazioni per il livello 1 ma chiede che si osservino i principi di buona sicurezza e igiene professionali.
(2) il D.lgs non parla nello specifico di autoclave ma richiede mezzi e procedure per il trattamento dei rifiuti.
l 06A P S A R A l Guida ai reparti per manipolazioni cellulari l Il concetto di contenimento
La cabina di sicurezza biologica
Le cabine di sicurezza biologica, dette Classe II
anche secondo la vecchia definizione Oltre a proteggere l’operatore e
cappe Biohazard, vengono solitamente l’ambiente, le cabine di classe II
classificate secondo la normativa proteggono anche il prodotto
americana NSF (National Sanitation manipolato. Il filtro HEPA è collocato
Foundation) in diverse classi a seconda sia sull’aria espulsa sia sull’aria che
delle caratteristiche tecniche che le ricircola all’interno della cabina
rendono adatte a diverse esigenze realizzando un flusso laminare sul
come schematizzato in tabella 3 dove piano di lavoro. Sono le cabine di
per completezza si riporta la vecchia sicurezza biologica più usate e sono
classificazione NSF49: 1992 e quella suddivise in 4 diversi tipi che si
attualmente in vigore NSF49: 2002. differenziano principalmente per la
percentuale di aria ricircolata rispetto
Classe I a quella espulsa.
Le cabine di classe I garantiscono il
ricircolo dell’aria nello spazio definito
dalla cabina stessa e la sua espulsione
in ambiente tramite un filtro tipo
HEPA.
Garantiscono la protezione dell’operatore
e dell’ambiente circostante la cabina,
ma non del prodotto manipolato.
NSF49:1992 NSF49:2002 Filtrazione HEPA Protezione Tabella 3
Classificazione
Classe Tipo Classe Tipo Ingresso Uscita Operatore Ambiente Prodotto cabine di sicurezza
I - I - NO SI buona ottima scarsa biologica
II A II A1 SI SI
II B1 II B1 SI SI buona ottima
II B2 II B2 SI SI
II B3 II A2 SI SI
III - III - SI SI (doppio) ottima ottima buona
l 07A P S A R A l Guida ai reparti per manipolazioni cellulari l Il concetto di contenimento
Figura 3
Cabina di sicurezza
biologica,
classe II tipo B1
Classe III
Tale cabina, realizza il massimo sull’aria in uscita, un passa materiali
contenimento ed è impiegata per la dedicato e il piano di lavoro
manipolazione di agenti biologici ad accessibile solo tramite guanti.
alto rischio. Sono previsti 2 filtri HEPA
Figura 4
Cabina di sicurezza
biologica, classe III
l 08A P S A R A l Guida ai reparti per manipolazioni cellulari l Il concetto di Cleanroom
Il concetto di cleanroom
In generale per cleanroom o camera bianca o ambiente a contaminazione
controllata si intende un ambiente in cui è possibile controllare la quantità
di polveri e mantenere la temperatura e l’umidità intorno a dei valori prefissati
allo scopo di proteggere il prodotto. Vengono utilizzate principalmente
dall’industria farmaceutica, medicale, alimentare e dall’industria elettronica.
Figura 5
Concetto di
cleanroom
Le cleanroom vengono suddivise in classi di contaminazione, secondo la quantità
di particelle per unità di volume; Questo tipo di classificazione corrisponde
allo standard definito dalle ISO EN 14644-1 che hanno sostituito le Federal
Standard 209 A che per prime, nel 1966, hanno definito le diverse classi di
contaminazione.
Nel caso di utilizzo nel settore farmaceutico, la normativa di riferimento è
rappresentata dalle Good Manufacturing Practice (GMP) per cui si ha una diversa
classificazione che tiene conto non solo delle particelle inerti come la classifica-
zione ISO, ma anche dei microrganismi teoricamente in grado di formare
colonie (CFU, dall’inglese colony forming units).
Il confronto delle condizioni “at rest” è rappresentato nella seguente tabella:
CLASSIFICAZIONE CLEANROOM Tabella 4
Confronto EU-GMP/Annex 1 e ISO 14644-1 (particelle/m3) Classificazione ISO
EU-GMP/Annex 1 ISO 14644-1 delle cleanroom
At rest
Grado 0.5µm 5µm 0.5µm 5µm Classe
A 3.500 1 3.520 29 ISO 5
B 3.500 1 3.520 29 ISO 5
C 350.000 2.000 352.000 2.930 ISO 7
D 3.500.000 20.000 3.520.000 29.300 ISO 8
l 09A P S A R A l Guida ai reparti per manipolazioni cellulari l Il concetto di Cleanroom
Le Good Manufacturing Practise (GMP)
Le Good Manufacturing Practice, Le classi di pulizia
in italiano Norme di Buona Il documento EC guide to good
Fabbricazione, sono il sistema di manufacturing practice revision to
qualità caratteristico dell’industria Annex 1 “Manufacture of sterile
farmaceutica. Stabiliscono una serie Medicinal Products” classifica
di regole che riguardano gli ambienti gli ambienti di manufacturing
e la strumentazione, i controlli secondo 4 gradi:
e il rilascio dei prodotti, lo stoccaggio grado A: zona di operazioni ad alto
delle materie prime e del prodotto rischio (riempimento, prodotto
finito per assicurare la qualità a contatto con l’ambiente). Il grado A
del prodotto finale in termini viene ottenuto mediante stazioni a
di caratteristiche fisico-chimiche flusso laminare che permettano
e di sicurezza per l’utente finale. l’ottenimento di una velocità dell’aria
Fino a pochi anni fà le GMP erano pari a 0,36-0,54 m/s sulla zona di
oggetto esclusivo dell’interesse lavoro. La maggior parte delle cabine
dell’industria farmaceutica di sicurezza di classe II in commercio
tradizionale. Attualmente il D.Lgs soddisfano tali requisiti.
n°211 del 24 giugno 2003, che recepi- grado B: per preparazioni asettiche
sce la direttiva europea 2001/20/CE, e operazioni di riempimento,
ha chiarito la necessità delle GMP già costituisce il background delle zone di
dalla fase di sperimentazione clinica. grado A.
La conoscenza e l’applicazione delle grado C e D: area per le fasi meno cri-
Norme di Buona Fabbricazione è tiche dei processi di produzione
quindi necessaria per tutti coloro che di prodotti sterili quali ad esempio
hanno superato la fase di ricerca e soluzioni successivamente sterilizzate
iniziano quella di sperimentazione per filtrazione.
nel settore della terapia cellulare e Vengono poi definite le seguenti
Figura 6 Le GMP della terapia genica. condizioni di lavoro:
at rest: impianti funzionanti,
strumentazione accesa, assenza
di personale
in operation: impianti funzionanti,
strumentazione accesa, presenza
di personale
l 10A P S A R A l Guida ai reparti per manipolazioni cellulari l Il concetto di Cleanroom
Grado At rest In operation Tabella 5
N° di particelle/m3 N° di particelle/m3 Limiti
monitoraggio
0,5 m 5m 0,5 m 5m
particellare
A ≤3.500 ≤1 ≤3.500 ≤1
B(1) ≤3.500 ≤1 ≤350.000 ≤2.000
C(1) ≤350.000 ≤2.000 ≤3.500.000 ≤20.000
D(1) ≤3.500.000 ≤20.000 non definito (2) non definito (2)
Oltre ai limiti illustrati nella tabella (1) Per raggiungere i gradi B, C e D il n° di ricambi aria/ora
precedente, sono di fondamentale necessario deve essere correlato alle dimensioni del
locale, alla strumentazione e al n° di persone presenti.
importanza i limiti relativi
Per i gradi A, B e C devono essere previsti appropriati
al monitoraggio microbiologico filtri terminali tipo HEPA.
che sono da intendersi in operation. (2) I limiti dipendono dalla natura delle operazioni svolte.
Tabella 6
Grado Campioni aria Settle plates Settle plates Guanto 5 dita
Limiti monitoraggio
cfu/m3 (Ø=9mm) cfu/4 (Ø=55mm) cfu/4 cfu/guanto microbiologico
AA P S A R A l Guida ai reparti per manipolazioni cellulari l Il concetto di Cleanroom
Il controllo ambientale della Cleanroom
Per mantenere la cleanroom nelle Monitoraggio dell’impianto
condizioni di progetto e verificare di condizionamenento
costantemente che tali condizioni Senza entrare nei dettagli tecnici,
siano mantenute durante il lavoro che esulano dallo scopo di questa
quotidiano degli operatori guida, la seguente tabella illustra
è indispensabile un sistema i parametri tipicamente monitorati
di controllo ambientale. in un impianto di condizionamento.
Oltre a monitorare i parametri tipici In molti caso al monitoraggio
che definiscono il corretto di un parametro segue un controllo
funzionamento degli impianti del parametro stesso e quindi
(es. temperatura, differenza l’intervento di un sistema
di pressione…), è possibile di supervisione per ripristinare,
monitorare altri parametri di più se possibile, le condizioni ambientali
specifico interesse per le cleanrooms. entro i limiti stabiliti.
Qualunque sistema usato per Opportuni riporti di allarme vengono
controllo o misurazione deve essere di norma previsti negli ambienti
sottoposto a verifica e calibrazione per informare tempestivamente
e tutte le attività di monitoraggio e gli operatori di condizioni fuori
controllo ambientale devono essere specifica.
documentate, registrate e conservate.
Tutti i sistemi di controllo devono
essere periodicamente verificati
per garantire affinché funzionino
in maniera adeguata.
Oggetto del monitoraggio Parametro controllato
Intasamento filtri
Macchine di condizionamento
Funzionamento macchina
(unità di trattamento aria, espulsori)
Temperature/portate fluidi a servizio delle macchine
Temperatura
Ambiente condizionato Umidità
Pressione differenziale tra diversi locali
Tabella 7 Monitoraggio impianto di condizionamento
l 12A P S A R A l Guida ai reparti per manipolazioni cellulari l Il concetto di Cleanroom
Monitoraggio particellare Monitoraggio microbiologico
Il monitoraggio particellare, come Il monitoraggio microbiologico
quello microbiologico, deve essere permette di verificare il numero
adeguato alle condizioni richieste di particelle vive presenti in ambiente
dalla classificazione ambientale e sulle superfici (mediante le
(cfr. tabella 5 e 6). cosiddette contact plates).
Il monitoraggio particellare può Ai sistemi tradizionali costituiti
essere eseguito con sistemi portatili da piastre con terreno di coltura
o con sistemi fissi in ambiente esposte all’aria o messe a contatto
in continuo. In entrambi i casi con le superfici da monitorare si sono
una quantità definita di aria viene recentemente affiancati sistemi
aspirata da una pompa e quindi che permettono campionamenti
un contatore di particelle laser conta multipli sequenziali dell’aria.
il numero delle particelle di una certa
dimensione rilevata. La posizione
e il numero di punti da monitorare
in ambiente è definito da specifici
documenti EN/ISO.
Questo tipo di monitoraggio non
distingue il tipo di particella (inerte
o microrganismo) presente in
ambiente.
In classe A, in accordo con la EC
guide to good manufacturing
practice revision to Annex 1
“Manufacture of sterile Medicinal
Products”, parte generale, nota a,
il monitoraggio particellare
in continuo viene richiesto per la
classe A (cappa) e raccomandato
per la classe B.
l 13A P S A R A l Guida ai reparti per manipolazioni cellulari l U n a m a n i p o l a z i o n e, u n a m b i e n t e
Una manipolazione, un ambiente
Nel momento in cui si deve allestire un’area dedicata alla terapia cellulare
o alla terapia genica, la normativa di riferimento può risultare a prima vista
complessa soprattutto a coloro che provengono dal mondo accademico
e dalla ricerca di base.
l 14A P S A R A l Guida ai reparti per manipolazioni cellulari l U n a m a n i p o l a z i o n e, u n a m b i e n t e
Quale ambiente scegliere
La seguente tabella schematizza, dato eseguire, il tipo di ambiente richiesto. Tabella 8
Una manipolazione,
lo scopo e il tipo di manipolazione da
un ambiente
Scopo della manipolazione cellu- Virus Tipo di ambiente GMP
si (wild type) contenimento no
Ricerca di base/ricerca preclinica si (MOGM) contenimento no
no contenimento no
Terapia genica si (MOGM) cleanroom + contenimento (1)
Impiego clinico Terapia cellulare no cleanroom (1)
NOTE(1) dipende dal tipo di manipolazione, vedi in particolare:
- Linee guida dell’Istituto Superiore di Sanità, Notiziario dell’Istituto (vol 7/8 luglio 2004)
- Conferenza stato-regioni 10 luglio 2003. Linee guida in tema di raccolta, manipolazione e impiego clinico delle cellule staminali emopoietiche /CSE
- Direttiva 2006/86/2006/CE. Attuazione direttiva 2004/23/CE del Parlamento europeo e del Consiglio per quanto riguarda le prescrizioni in tema
di rintracciabilità, la notifica di reazioni ed eventi avversi gravi e determinate prescrizioni tecniche per la codifica, la lavorazione, la conservazione,
lo stoccaggio e la distribuzione di tessuti e cellule umane
- DM 5/12/2006. Utilizzazione di medicinali per terapia genica e per terapia cellulare somatica al di fuori di sperimentazioni cliniche e norme
transitorie per la produzione di detti medicinali
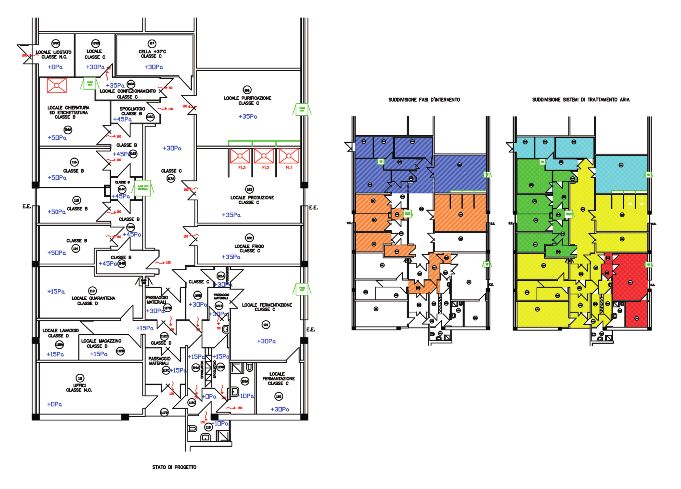
Esempi di layout
Di seguito sono mostrati alcuni esempi e laboratori classificati secondo GMP.
di layout di laboratori a contenimento
Figura 7
Esempio layout
laboratorio PCL2
l 15A P S A R A l Guida ai reparti per manipolazioni cellulari l U n a m a n i p o l a z i o n e, u n a m b i e n t e Figura 8 Esempio layout laboratorio PCL3 Figura 9 Esempio layout laboratorio GMP l 16
A P S A R A l Guida ai reparti per manipolazioni cellulari l Glossario
Glossario
A E I
Agente > Qualsiasi potere, principio, Efficienza filtri ISO
sostanza, biologico, chimico o fisico Rapporto tra la differenza della con- International Organization for
in grado di agire su un organismo centrazione di particelle monte/valle Standardization
del filtro e quella
a monte. M
MOGM
B F Micro Organismo Geneticamente
Biohazard Filtro a carbone attivo Modificato
Contrazione della parola Biological e Filtro specifico ad alto adsorbimento
Hazard. per il trattamento di microparticelle
Biosafety cabinet volatili, usato per il controllo della N
Termine inglese, tipicamente usato contaminazione chimica spesso a NIH
negli Stati Uniti, equivalente a cabina valle di filtri HEPA. Acronimo di National Institute of
di sicurezza biologica ( vedi Cabina Filtro Assoluto Health, ente americano equivalente
di Sicurezza Biologica). Termine obsoleto per indicare il filtro all’italiano Istituto Superiore di
BL (1, 2, 3 o 4) HEPA. Sanità
Acronimo di Biosafety Level secondo Filtro HEPA NSF
le linee guida del NIH (( vedi NIH) High Efficiency Particulate Air Filter. Acronimo di National Sanitation
Filtro tipicamente in microfibra di Foundation, ente americano no
borosilicato, pieghettato e montato profit di elaborazione di standards
su telaio con capacità di rimozione di
particelle di 0,3(m (efficienza
C di rimozione) minima dell’85% P
Cabina di Sicurezza Biologica (tipo H10) e massima del 99,995% PCL (1, 2, 3 o 4)
Cabina a flusso laminare che, (tipo H14). Acronimo di Phisycal Containment
mediante il combinato meccanismo Sono usati per rimuovere il articolato Level secondo lo standard ISO
della ventilazione dall’aria, non sono utili in caso di gas (vedi ISO) EN12128.
e di filtrazione dell’aria, costituisce o vapori nocivi.
l’elemento di contenimento primario Flusso Laminare
della dispersione dei contaminanti. Flusso d’aria che all’interno di uno S
Al flusso laminare di aria si associa spazio definito si muove con velocità SOP
un secondo flusso d’aria che entra uniforme lungo linee parallele. Acronimo di Standard Operating
attraverso l’apertura di accesso for- Termine che tende ad essere Procedure, in italiano procedura
mando la barriera di protezione sostituito nei nuovi documenti operativa standard. Definisce
a cui corrisponde un uguale normativi con la dizione nei dettagli come effettuare una certa
flusso d’aria uscente. “flusso unidirezionale”. operazione.
Camera bianca
Vedi Clean Room. G
Cappa biohazard a flusso laminare Glove box U
Vecchia definizione dell’attuale cabina In italiano letteralmente “scatole a UNI
di sicurezza biologica. guanti” sono sistemi caratterizzati Ente Nazionale Italiano di
CFU da chiusura ermetica rispetto Unificazione, partecipa per l’Italia
Acronimo di Colony Forming Units, all’ambiente esterno. Termine spesso all’attività normativa degli organismi
in italiano unità formanti colonie. usato impropriamente come sinonimo sovranazionali di normazione: ISO
cGMP di cabina di sicurezza biologica di (International Organization for
Acronimo di current Good tipo III. Standardization) e CEN (Comité
Manufacturing Practice GMP Européen de Normalisation).
Classificazione filtri Good Manufacturing Practice,
I filtri sono classificati secondo la loro in italiano Norme di Buona
efficienza di filtrazione. Fabbricazione. Dato che le norme V
Si distinguono 4 classi. sono in continua evoluzione di parla Validazione
I più conosciuti in ambiente fa spesso di cGMP (current Good Serie di operazioni attraverso cui
rmaceutico sono i filtri HEPA Manufacturing Practice) si giudica lo scarto tra i risultati attesi
(vedi filtro HEPA) di una misura, di un test o
Clean room H di un processo e i risultati misurati.
Ambiente con numero controllato di HEPA∞ Tale scarto deve rimanere entro limiti
particelle di dimensioni note. Vedi filtro HEPA predeterminati.
l 17A P S A R A l Guida ai reparti per manipolazioni cellulari l Documentazione di riferimento
Elenco documentazione di riferimento
Nei paragrafi seguenti vengono indicati i principali documenti di riferimento. Nell’ordine vengono indicati i decreti legge,
le circolari e le linee guida italiane, le direttive e tutti gli altri documenti a livello europeo e quindi quelli americani.
SICUREZZA / RISCHIO BIOLOGICO
D.Lgs. n°626 del 19 settembre 1994 integrato con D.Lgs. n°242 del 18 marzo 1996. Miglioramento della sicurezza
e della salute dei lavoratori sul luogo di lavoro
Decreto Lgs. N°206 12 aprile 2001. Attuazione della direttiva 98/81/CE che modifica la direttiva 90/219/CE,
concernente l’impiego confinato di microrganismi geneticamente modificati
UNI EN 12128, 2000. Laboratori di ricerca, sviluppo, analisi. Livelli di contenimento di laboratori microbiologici,
aree di rischio, situazioni e requisiti di sicurezza
Primary Containment for Biohazards. Selection, Installation and Use of Biological Safety Cabinets 2nd edition,
2000 U.S. Government printing office
Laboratory Biosafety Manual (Organizzazione Mondiale Sanità) 3rd edition, 2004
GMP SPERIMENTAZIONE CLINICA
DM 5/12/2006. Utilizzazione di medicinali per terapia genica e per terapia cellulare somatica al di fuori
di sperimentazioni cliniche e norme transitorie per la produzione di detti medicinali
Direttiva 2006/86/2006/CE. Attuazione direttiva 2004/23/CE del Parlamento europeo e del Consiglio per quanto
riguarda le prescrizioni in tema di rintracciabilità, la notifica di reazioni ed eventi avversi gravi e determinate
prescrizioni tecniche per la codifica, la lavorazione, la conservazione, lo stoccaggio e la distribuzione di tessuti
e cellule umane
Direttiva Europea 2004/23/CE. Definizione di qualità e di sicurezza per la donazione, l’approvvigionamento,
il controllo, la lavorazione, la conservazione, lo stoccaggio e la distribuzione di tessuti e cellule umane
D.Lgs. n°211 24 giugno 2003. Attuazione della direttiva 2001/20/CE relativa all’applicazione della buona pratica clinica
nell’esecuzione delle sperimentazioni cliniche di medicinali per uso clinico
D.Lgs. n°178 29 maggio 1991. Recepimento delle direttive delle comunità economica europea in materia di specialità
medicinali
DPR 439/2001 del 21 settembre 2001. Regolamento di semplificazione delle procedure per la verifica e il controllo
di nuovi sistemi e protocolli terapeutici sperimentali
EC Guide to Good Manufacturing Practice Annex 1, 2003
EC Guide to Good Manufacturing Practice Annex 2. Manufacture of Biological Medicinal Products for Human Use
Vol.4 Good Manufacturing Practice Annex 13. Manufacture of Investigational Medicinal Products, 2003
Vol.4 EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, 2005
ISO 14664-1, 1999 Cleanrooms and associated controlled environments Part 1. Classification of airbone particulates
ISO 14664-2, 2000 Cleanrooms and associated controlled environments Part 2. Specifications for testing and
monitoring to prove continued compliance with ISO 14644-1
ISO 14664-5, 2004 Cleanrooms and associated controlled environments Part 5. Operations
Notiziario dell’Istituto Superiore di Sanità vol. 7/8 luglio 2004
AIFA- documento della qualità. Linea guida per le ispezioni ai produttori di medicinali per terapie avanzate
e per terapia cellulare somatica, DSQ/12 rev0 del 5 marzo 2007
Conferenza stato-regioni 10 luglio 2003. Linee guida in tema di raccolta, manipolazione e impiego clinico delle cellule
staminali emopoietiche /CSE
Guidance for Industry, 2004. Sterile Drug Products Produced by Aseptic Processing- Current Good Manufacturing
Practice
Guidance for Industry, 2001. Q7A Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients
Guideline on human cell-based medicinal products, DRAFT, dicembre 2006 EMEA/CHMP/410869/2006
l 18Referenze Apsara
l 19A P S A R A l Guida ai reparti per manipolazioni cellulari l Referenze
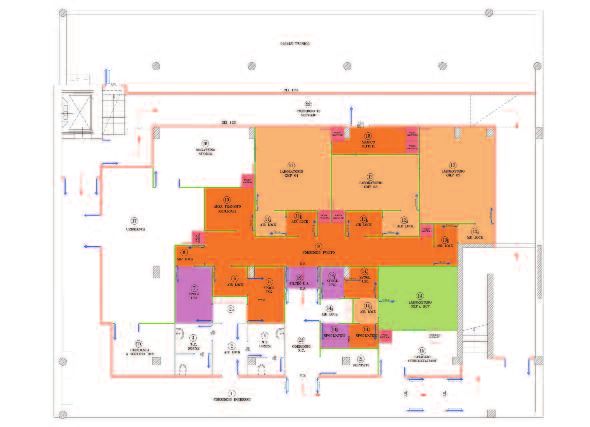
ARETA INTERNATIONAL - Insubria Biopark Varese
Laboratori GMP “Clinical Grade”
NUOVO OSPEDALE DI MESTRE
Laboratori GMP Banca dell’Occhio
l 20A P S A R A l Guida ai reparti per manipolazioni cellulari l Referenze
MOLMED SPA Milano
Laboratori GMP Terapie Innovative
UNIVERSITA’ DI TORINO
Laboratori GMP Scuola di Biotecnologie
l 21A P S A R A l Guida ai reparti per manipolazioni cellulari l Referenze
BIOSCIENCE INSTITUTE SAN MARINO
Laboratori GMP Cell Factory
AZIENDA OSPEDALIERO UNIVERSITARIA PISANA
Laboratori GMP e Banca dei tessuti
l 22Puoi anche leggere

















































