Bioactive molecule How design an in vivo - Metodologie di Sintesi e Sviluppo Farmaceutico - E-learning
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Metodologie di Sintesi e Sviluppo Farmaceutico
Synthesis and Development Pharmaceutical Methodologies
Laurea Magistrale in Chimica a.a. 2018/2019
How design an in vivo
bioactive molecule
Marco L. Lolli University of Torino (UniTO)
FASI DELL’AZIONE DI UN FARMACO
Compresse,
capsule,sciroppo, supposte,
Forma Farmaceutica
preparazioni iniettabili, etc.
FASE Somministrazione
FARMACEUTICA Liberazione dalla forma farmaceutica
Passaggio in soluzione
FASE Assorbimento
FARMACOCINETICA Distribuzione ADME
Metabolismo
Escrezione
FASE Interazione Biofase
FARMACODINAMICA Farmaco-sito d’azione
Effetto
Marco L. Lolli University of Torino (UniTO)
Pharmacokinetics (ADME)
“The movement of a drug through the body”
Adorption
Distribution
Metabolism
Elimination
Tox
Marco L. Lolli University of Torino (UniTO)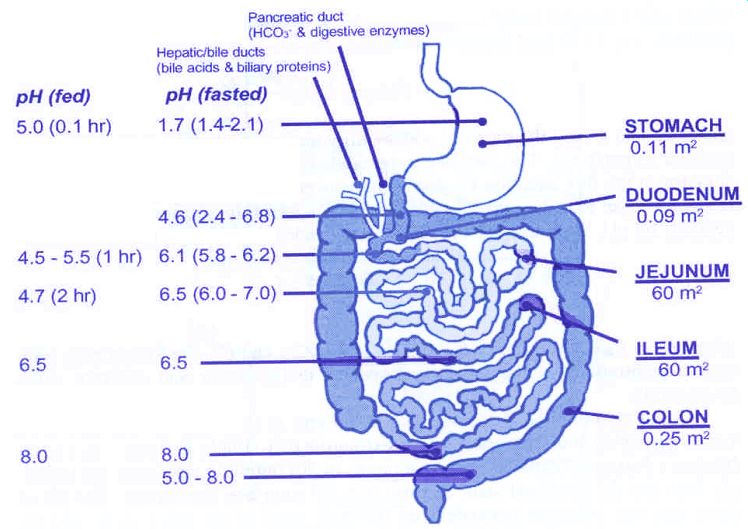
Druggability
Ligand side
The Lipinski ‘’rule of five’’
In the original Lipinski paper was started hat poor absorption or
permeation are more likely when:
1) In the structure are present more than 5 hydrogen-bond
donors (HBD).
2) The MWT is over 500 Daltons
3) The LogP is over 5 (or MLogP is over 4.15).
4) There are more than 10 H-bond acceptors.
5) Substrates for biological transporters are exceptions to the
rule.
Lipinski CA, Lombardo F, Dominy BW, Freeney PJ. Experimental and computational approaches to estimate solubility and
per- meability in drug discovery and development settings. 1997. Adv Drug Deliv Rev 23, 3 - 25.
Marco L. Lolli University of Torino (UniTO)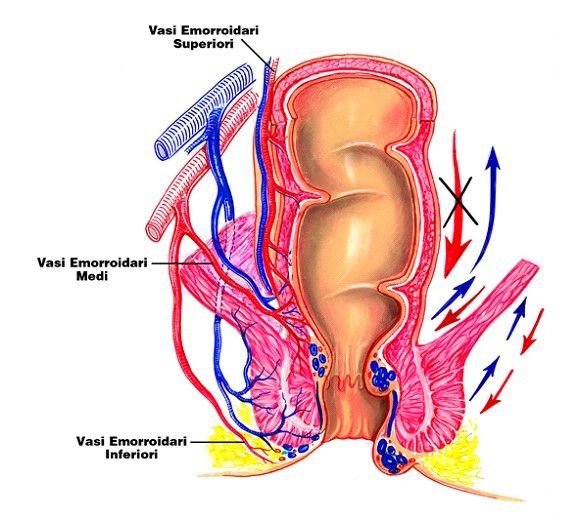
FARMACOCINETICA: ciò che l’organismo fa al farmaco
Distribuzione
F-P
Assorbimento F Escrezione
metaboliti
sangue
Metabolismo
Studia il movimento dei farmaci nell’organismo ed i processi che
determinano la quantità di farmaco disponibile al sito di azione
Marco L. Lolli University of Torino (UniTO)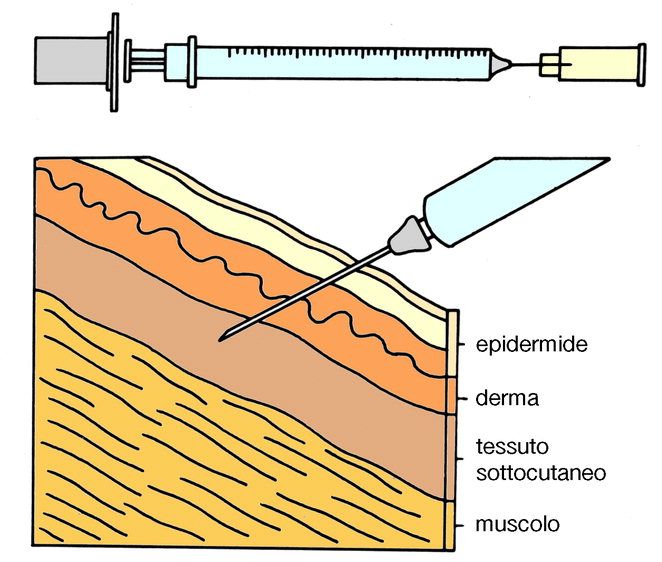
Assorbimento: ADME
Passaggio del farmaco nel torrente circolatorio
q formulazione
q via di somministrazione (e.v.: l’assorbimento non è necessario)
q presenza di altri farmaci o alimenti
q proprietà chimico-fisiche del farmaco:
v solubilità
v grado di ionizzazione
v lipofilia
v dimensione molecolare
Marco L. Lolli University of Torino (UniTO)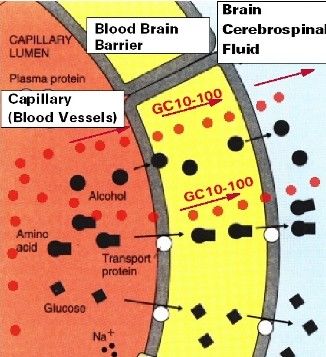
Nel suo viaggio attraverso il corpo un farmaco deve attraversare
diverse barriere biologiche.
Queste barriere possono essere costituite da:
- parecchi strati di cellule (pelle)
- un singolo strato di cellule (l’epitelio intestinale)
- la membrana cellulare stessa (per raggiungere
un recettore intracellulare).
Marco L. Lolli University of Torino (UniTO)
Forme Farmaceutiche
Possono essere suddivise in accordo alla loro natura fisica in:
Principio attivo
+
1. Liquide: soluzioni
Eccipienti:
sospensioni
• Riempienti
emulsioni
• Lubrificanti
2. Semisolide: creme
• Leganti
unguenti
• Disintegranti
gel
• Conservanti
3. Solide: capsule
• Antiossidanti
compresse
• Modificatori del gusto
supposte, ovuli
Marco L. Lolli University of Torino (UniTO)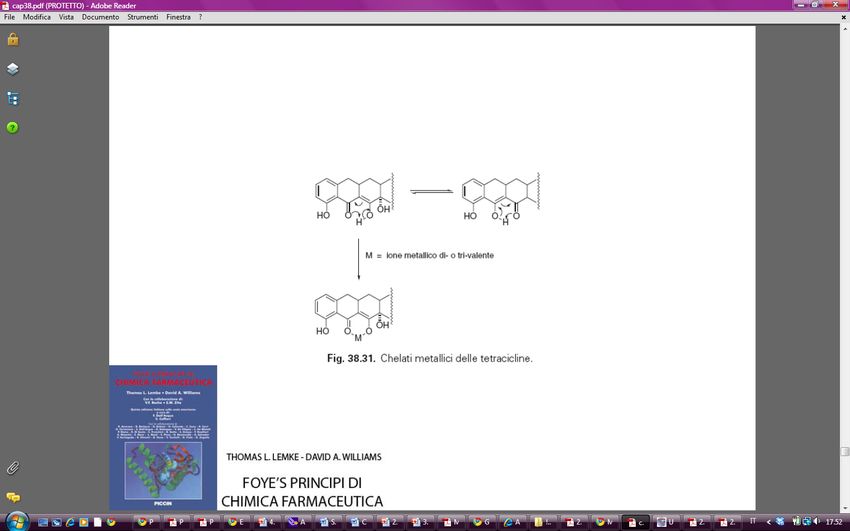
Biodisponibilità: ADME
frazione della dose somministrata che raggiunge la circolazione
sistemica non modificata.
Dipendente da:
• Modalità di somministrazione
• Metabolismo
Marco L. Lolli University of Torino (UniTO)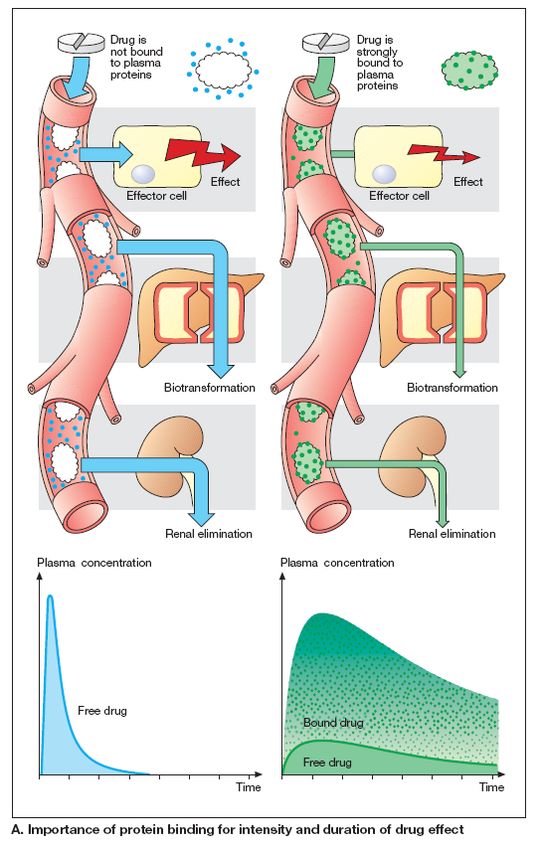
Vie di Somministrazione
8
1. Topico
4 1, 2
2. Sistemico
A. Enterale
1. orale (per os)
2. sublinguale 7
3. rettale 6
B. Parenterale (evita il sist. GI)
4. iniezione endovenosa (e.v.)
5. iniezione intramuscolare (i.m.) 5
6. iniezione sottocutanea (s.c.)
7. transdermica
8. inalatoria
3
Marco L. Lolli University of Torino (UniTO)Somministrazione Topica
Applicazione locale esterna (es. pelle, mucose, parti del corpo poco
irrorate) quando l’assorbimento sistemico NON è desiderabile, per
esplicare effetti terapeutici in queste sedi (antiinfettivi, antiinfiammatori).
Antiacidi, antidiarroici, spasmolitici, e antisettici intestinali sono assunti
per via orale per svolgere un’azione circoscritta al tratto
gastrointestinale e grazie alla loro scarsa biodisponibillità orale (troppo
idrofili o poco solubili) non hanno effetti sistemici.
Anche la via polmonare può essere utilizzata per somministrazioni
topiche (broncodilatatori antiasmatici).
Marco L. Lolli University of Torino (UniTO)Somministrazione Sistemica
Se il farmaco viene assorbito ed entra in circolo .
Enterale se attraversa il sistema gastroenterico,
Parenterale se lo evita.
La via dipende dalla stabilità chimica del farmaco, dalla sua capacità
di attraversare le membrane e arrivare al sito d’azione, dall’età del
paziente e dalle capacità fisiche e mentali del paziente.
Marco L. Lolli University of Torino (UniTO)Enterale Enterale
Ogni tratto differisce per il pH, per gli enzimi
presenti, per la superficie di assorbimento, per la
fluidità dei contenuti.
Il tratto in cui il farmaco viene maggiormente
assorbito è il duodeno e il tenue, perché qui la
parete è più estesa (villi intestinali), sottile e
permeabile (200 mq di epitelio monostratificato
contro 1 mq pluristratificato dello stomaco).
Infine la perfusione ematica dell’intestino è 7
volte quella dello stomaco.
Marco L. Lolli University of Torino (UniTO)Per OS Enterale
E’ una delle vie più comuni e costituisce la via di prima
scelta per la somministrazione agli adulti per l’elevata
compliance.
Efficace per farmaci stabili a pH acido ed agli enzimi
digestivi che possono essere formulati in capsule,
pillole o soluzioni (maggior assorbimento per farmaci).
Per capsule o polvere la velocità dipende dalla velocità
di dissoluzione legato alle dimensioni delle particelle e
alle forme cristalline.
Pillole o caspule progettate in modo speciale possono
restare intatte nello stomaco, per aiutare a proteggere
farmaci acido-labili dagli acidi dello stomaco; gli
involucri poi si degradano una volta arrivati
nell’intestino.
Marco L. Lolli University of Torino (UniTO)Per OS: Influenza del pH Marco L. Lolli University of Torino (UniTO)
Per OS: Influenza del pH
In assenza di carrier specifici, la forma che oltrepassa più facilmente le membrane lipidiche in
virtù di una maggiore lipofilia è la forma non-ionizzata:
• I farmaci con proprietà acide sono maggiormente assorbiti a livello dello stomaco (pH 2-3)
• I farmaci con proprietà basiche sono maggiormente assorbiti a livello dell’intestino (pH 5-7).
Aspirine Amphetamine
Marco L. Lolli University of Torino (UniTO)Dimensioni
C6H12N4 C10H16 C10H16
PM ≠; dimensioni ≅ PM =; dimensioni ≠
Diffusione paracellulare
Diffusione passiva
Velocità:
OH NH3+ inversamente proporzionale al PM
dC D A . P (Cout – Cin)
.
- =
dt h
Marco L. Lolli University of Torino (UniTO)Dimensioni
ATTRAVERSANO LA MEMBRANA CELLULARE SENZA TRASPORTATORE
Ø MOLECOLE LIPOFILE (logP > 0) DIFFUSIONE PASSIVA
Ø ZWITTERIONI (logP > -2) DIFFUSIONE PASSIVA
Ø SOSTANZE DI PICCOLISSIME DIMENSIONI: PM < 150 Da DIFFUSIONE CONVETTIVA
NON ATTRAVERSANO LA MEMBRANA CELLULARE SENZA TRASPORTATORE
Ø SOSTANZE IDROFILE con logP < 0 e PM > 150 Da
Ø SOSTANZE TROPPO LIPOFILE: logP > 6
Ø SOSTANZE TROPPO VOLUMINOSE: PM > 1000 Da (proteine, acidi nucleici)
L’identikit di oltre il 90% dei farmaci: ü PM < 500 Da
ü diametro < 20 Å
ü neutro o ionizzabile
ü logP < 5
Marco L. Lolli University of Torino (UniTO)Assorbimento e grado di ionizzazione
Farmaci neutri
DIFFUSIONE PASSIVA
Neutro Neutro non reagiscono con H2O né come acidi né come basi
Acidi e basi deboli
Ionizzato Ionizzato
In soluzione acquosa sono coinvolti in
equilibri in cui la forma neutra coesiste
con la forma ionizzata, dotata di carica NH3+
negativa o positiva
Marco L. Lolli University of Torino (UniTO)Equazione di Henderson-Hasselbalch
Marco L. Lolli – Faculty of Pharmacy – University of Turin
(percentuale di ionizzazione)
[A-][H+] [A-]
Ka = pH = pKa + log
[HA] [HA]
Per un acido Per una base
2.8 3.8 4.8 5.8 6.8 2.8 3.8 4.8 5.8 6.8
HA 100 90 50 10 1 BH+ 100 90 50 10 1
A- 0 10 50 90 100 B 0 10 50 90 100
Marco L. Lolli University of Torino (UniTO)DIFFUSIONE PASSIVA Assorbimento e grado di ionizzazione
ACIDI E BASI DEBOLI
Neutro Neutro
100
% dissociazione =
1 + 10 (pKa – pH)
Ionizzato Ionizzato
pKa N.B.: farmaci come acidi
3-5
acidi carbossilici
0-1
- acidi solfonici
AH A- + H+
9-10
+
amine BH+ B: + H+
10-11
amidine
12-13 acido forte
acidi: valore di pKa basso
guanidine basi: valore di pKa alto base forte[A-] [H+]
Per gli acidi deboli: AH A- + H+ Ka = pKa = -log Ka
[AH]
Grado di dissociazione
molecole dissociate
molecole dissociate
α= molecole totali
[A-] dividendo tutto
% dissociazione = . 100 per [A-]
[AH] + [A-]
molecole totali
100 [AH] [H+]
% dissociazione = dalla definizione di = sostituendo
[AH] Ka [A-] Ka
+ 1
[A-]
100
% dissociazione = 100
[H+] % dissociazione =
1 + (pKa-pH)
Ka 1 + 10
[OH-] [BH+]
Per le basi: B + H 2O BH+ + OH- Kb = pKb = -log Kb
[B]
pKa + pKb = pKw = 14 Convenzionalmente
formulata come pKaAssorbimento e grado di ionizzazione
pKa = 7 pH = 7
pKa = 7 pH = 8 ≈
pKa = 7 pH = 6 ≈
Somministrazione orale: assorbimento attraverso l’epitelio gastrico e
l’epitelio intestinale
basi deboli: assorbimento attraverso l’epitelio intestinale
acidi deboli: il pH dello stomaco + favorevole
• tempo di permanenza limitato
• superficie relativamente piccola dello stomaco
maggiormente assorbiti attraverso l’epitelio intestinale
pH succo gastrico: 1-2
pH duodenale: 4 - 5
pH ileo: 6 - 8
mucosa intestinale: villi e microvilli , superficie ~250 m2
disponibile per la diffusione passivaAssorbimento e grado di ionizzazione Marco L. Lolli University of Torino (UniTO)
Assorbimento e grado di ionizzazione
N Limitato assorbimento
Stomaco, pH = 1 - 2
nello stomaco
(anatomia)
N N Buon assorbimento
nel duodeno
Duodeno, pH = 4 - 5
Buon assorbimento
N N nell’intestino tenue (digiuno + ileo)
Intestino tenue, pH = 6 - 8
sangue pH = 7.4
Marco L. Lolli University of Torino (UniTO)Assorbimento e grado di ionizzazione
ACIDO DEBOLE, pKa = 4
Limitato assorbimento
HA nello stomaco
Stomaco, pH = 1 - 2
(anatomia)
HA HA A- Buon assorbimento
nel duodeno
Duodeno, pH = 4 - 5
A-
HA HA A- Assorbimento molto più lento
nell’intestino tenue (digiuno + ileo)
Intestino tenue, pH = 6 - 8
A- sangue pH = 7.4
Marco L. Lolli University of Torino (UniTO)Assorbimento e grado di ionizzazione
BASE DEBOLE, pKa = 5
BH+ NO assorbimento
Stomaco, pH = 1 - 2 nello stomaco
B B BH+
Scarso assorbimento
nel duodeno
Duodeno, pH = 4 - 5
BH+
B B BH+ Buon assorbimento
nell’intestino tenue (digiuno + ileo)
Intestino tenue, pH = 6 - 8
BH+ sangue pH = 7.4
Marco L. Lolli University of Torino (UniTO)Assorbimento e grado di ionizzazione
BASE FORTE, pKa = 9
BH+
Stomaco, pH = 1 - 2 NO assorbimento
nello stomaco
B B BH+ NO assorbimento
nel duodeno
Duodeno, pH = 4 - 5
BH+
B B BH+ Scarso o moderato assorbimento
nell’intestino tenue (digiuno + ileo)
Intestino tenue, pH = 6 - 8
BH+ sangue, pH = 7.4
Marco L. Lolli University of Torino (UniTO)Assorbimento e grado di ionizzazione
SOSTANZE ANFOTERE: funzioni acide e basiche nella stessa molecola
Una sostanza anfotera può esistere in soluzione come zwitterione, una specie con due cariche locali di
segno opposto e una carica netta uguale a zero
punto isoelettrico (pI): pH al quale risulta massima la percentuale della forma zwitterionica
pKa1 + pKa2
pI =
2
pKa = 9.6
pIamoxicillina = (9.6 + 2.4)/2 = 6
pKa = 2.4
pH a cui oltre il 99,9% dell’amoxicillina si
trova all’equilibrio come zwitterione
pKa = 6.0
pIciprofloxacina = (6.0 + 8.8)/2 = 7.4
la specie zwitterionica prevale
pKa = 8.8 (> 50%) a valori di pH compresi tra
pKa1 e il pKa2
Marco L. Lolli University of Torino (UniTO)Assorbimento e grado di ionizzazione
Lo stato di zwitterione può essere vantaggioso per un farmaco:
§ privi di carica netta: gli zwitterioni riescono ad attraversare le membrane cellulari
Gli zwitterioni sono più idrosolubili
delle specie neutre e meno idrosolubili
delle specie ioniche (dotate di carica
netta)
S
COOH COO- COO-
NH3+ NH3+ NH2
pH iso pH
Marco L. Lolli University of Torino (UniTO)FARMACI ORGANICI CON CARICA PERMANENTE
Idrofili (log P < -3)
Sali d’ammonio quaternario
Molto solubili o solubilissimi in H2O
(So > 10% g/mL)
proprietà indipendenti dal pH
Ioscina butilbromuro (Buscopan®)
azione a livello del tratto GI
riduzione motilità gastrointestinale
Ø Non attraversano le membrane cellulari
Ø non sono generalmente assorbiti in seguito a somministrazione orale
Marco L. Lolli University of Torino (UniTO)Via Sublinguale Enterale
La cavità buccale è riccamente irrorata da vasi sanguigni e il composto passa direttamente nel
circolo sistemico. Adatta a farmaci molto lipofili, non adatta a tutti i farmaci od a farmaci da
somministrare ad alte dosi.
Es.:
trinitroglicerina per pazienti cardiopatici.
Fentanile (analgesico oppiaceo)
sottoforma di leccalecca ai bambini.
Cocaina, masticando le foglie di coca.
Marco L. Lolli University of Torino (UniTO)Via Rettale Enterale
I principi attivi vengono drenati dal plesso emorroidario inferiore; solo una piccola frazione passa per il
fegato, la maggior parte entra direttamente nel torrente circolatorio.
- Adatta a farmaci sufficientemente lipofili,
- Assorbimento rapido (il retto è molto vascolarizzato-plesso emorroidario inferiore).
- Adatta se il paziente ha vomito, è privo di coscienza o non può deglutire (pediatria).
• % assorbimento variabile
• scarsa compliance
• ~ 50% della dose soggetta alla
metabolizzazione di primo passaggio
Marco L. Lolli University of Torino (UniTO)Metabolismo di primo passaggio
Trasformazioni chimiche e metaboliche che avvengono prima che il farmaco
arrivi nella circolazione sistemica; farmaco completamente degradato dal
metabolismo di primo passaggio: non somministrabile per via orale.
Per evitare l’effetto di primo
passaggio e la conseguente
ridotta biodisponibilità è
possibile utilizzare vie di
somministrazione alternative
(sublinguale, rettale,
polmonare, transdermica)
Marco L. Lolli University of Torino (UniTO)Marco L. Lolli University of Torino (UniTO)
La biodisponibilità
è una misura della quantità di principio attivo che raggiunge il circolo sistemico ed è
disponibile presso il sito di azione per esercitare un effetto sul target biologico. Un effetto di
primo passaggio molto pronunciato, degradazione del principio attivo (ad esempio ad opera
dell’acidità dei succhi gastrici o di enzimi idrolitici) un cattivo assorbimento dovuto a una
formulazione inadeguata, possono dare origine a una scarsa biodisponibilità.
Marco L. Lolli University of Torino (UniTO)ABSORPTION 249
La biodisponibilità
(a) (b)
FIGURE 6.3 (a) The oral pharmacokinetic profile of a drug is determined by its absorp-
tion, distribution, metabolism, and excretion. (b) Oral delivery of a drug leads to an initial
rise in(a)plasma
The oral pharmacokinetic
concentration until profile of a drug
the maximum is determined
concentration by ) its
(Cmax absorption,
is reached (Tmax). When
distribution, metabolism, and excretion.
the rate of elimination becomes greater than the rate of absorption, plasma concentration
(b) Oral delivery of a drug leads to an initial rise in plasma concentration until the
decreases. When absorption is complete, the drug is removed through elimination pathways.
maximum concentration (Cmax) is reached (Tmax). When the rate of elimination
becomes greater than the rate of absorption, plasma concentration decreases. When
the absorption
body is complete,
(Figure the drug
6.3(a)). is removed
Oral throughof
delivery elimination
a drugpathways.
leads to an initial
rise in
plasma concentration as the drug is absorbed.
MarcoAs the drug isUniversity
L. Lolli distributed
of Torino (UniTO)
through the body, metabolism and excretion eliminate the drug from sys-La biodisponibilità
Endovena: biodisponibilità 100 %
Intramuscolo sottocutaneo
biodisponibilità ≅ 100 % o poco
inferiore
Per os: biodisponibilità 0 ÷ 100 %; la maggior
parte biodisponibilità > 20 %; se < 5
% azione localizzata nel tratto GI
Marco L. Lolli University of Torino (UniTO)Via Parenterale: iniezione
- Evita l’apparato gastroenterico. Risposta più rapida che per os.
- Livelli di farmaco somministrati sono più accurati.
- Pericolosità più elevata
(reazioni inaspettate, essenziale la sterilità, rischi di overdose).
- Socialmente avversa
Può essere:
• sottocutanea (SC)
• intramuscolo (IM)
• endovena (EV)
• intratecale (IT)
• intraperitoneale (IP)
Marco L. Lolli University of Torino (UniTO)Via Parenterale: iniezione sottocute ed intramuscolo
Per farmaci non irritanti i tessuti per evitare dolore
SC IM
intenso, necrosi e desquamazione.
• assorbimento veloce da soluzioni acquose
• assorbimento lento e costante da formulazioni
oleose o sospensioni
Pro:
• aggirato l’effetto di primo passaggio
• praticabili se il paziente non è cosciente
• assorbimento può essere rapido (i.m.)
• particolarmente adatte per forme ritardo
Contro:
• pericolosità intermedia tra la via orale e quella e.v.
• scarsa compliance
Marco L. Lolli University of Torino (UniTO)Via Parenterale: iniezione indovena
La più efficace per somministrare farmaci in dosi
accurate, ed anche la più veloce e la più pericolosa.
Le soluzioni devono essere:
• Limpide: solamente farmaci disciolti in soluzione.
• Acquose: farmaci solubili in liquidi oleosi non possono
essere somministrati e. v. perché potrebbero insorgere
emboli.
• Sterili
• Isotoniche
• pH molto vicino a quello del plasma (pH 7.4).
Scarsa compliance.
Marco L. Lolli University of Torino (UniTO)Via Transdermica
Farmaco a contatto con la cute (pomate, emulsioni, cerotti)
e viene assorbito nel torrente circolatorio a livello dell’area
di contatto.
Solo per farmaci lipofili (epidermide è una barriera lipidica).
Cerotti a rilascio controllato: il farmaco viene liberato
lentamente (alcuni giorni). I livelli plasmatici sono
relativamente costanti. Usato per nicotina, ormoni
per la terapia sostitutiva per l’estrogeno, fentanile
(analgesico) e clonidina (antiipertensivo).
Marco L. Lolli University of Torino (UniTO)Via inalatoria
Somministrazioni sistemiche:
per farmaci gassosi o volatili (anestetici generali) con
assorbimento rapido attraverso i capillari del
circolo polmonare.
Farmaci non gassosi possono essere somministrati
come aereosol.
Molte sostanze da abuso sono assorbite attraverso
questa via: nicotina, cocaina, marijuana,
metamfetamina, eroina.
Marco L. Lolli University of Torino (UniTO)Somministrazioni topiche per
patologie polmonari:
per evitare l’assorbimento si preferiscono
farmaci molto polari. Molto usato per i
broncodilatatori antiasmatici con indesiderati
effetti a livello cardiaco (aerosol, spray nasali).
Marco L. Lolli University of Torino (UniTO)Permeability:
Capacità di superare le barriere biologiche
q proprietà chimico-fisiche del farmaco (lipofilia, caratteristiche acido-base, PM)
q caratteristiche anatomo-fisiologiche dell’organo
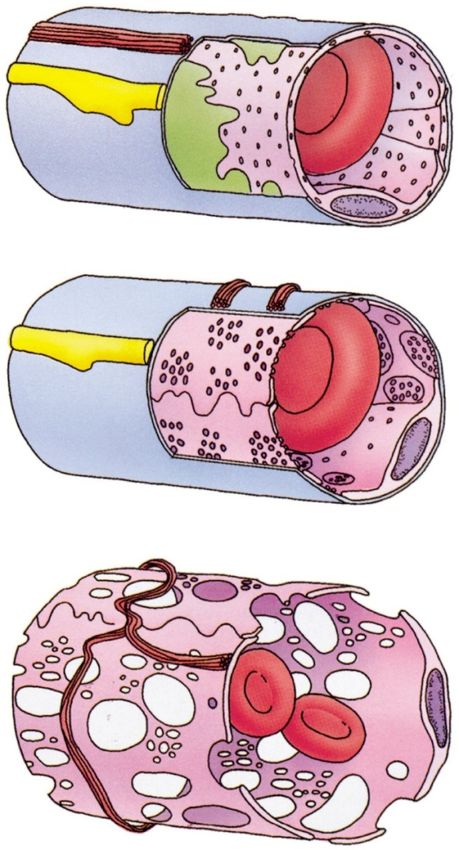
Marco L. Lolli University of Torino (UniTO)Nel suo viaggio attraverso il corpo un farmaco deve attraversare
diverse barriere biologiche.
I farmaci possono attraversare gli strati cellulari con due modalità generali:
diffusione paracellulare: passando attraverso gli interstizi acquosi tra le cellule
diffusione transcellulare: attraversando la cellula stessa.
Marco L. Lolli University of Torino (UniTO)compound permeability. Compounds intended to modulate CNS func-
tions, for example, must traverse the blood–brain barrier, which is far more
restrictive in nature. Tight cellular junctions and higher levels of transport
Nelprotein
suo viaggio attraverso il corpo un farmaco deve attraversare
expression designed to protect the brain from xenobiotics must be
diverse barriere
overcome biologiche.
in order for CNS targeted compounds to elicit a response.
The observed permeability of any given compound across a biologi-
cal barrier is the sum of five modes of transport: passive diffusion, active
transport, endocytosis, paracellular transport, and efflux (Figure 6.10 ). In
(a) (b) (c) (d) (e)
FIGURE 6.10 (a) Passive diffusion; (b) Active transport; (c) Endocytosis; (d) Efflux; (e)
Paracellular transport.
(a) Passive diffusion; (b) Active transport; (c) Endocytosis; (d) Efflux; (e) Paracellular transport.
considering the transfer of potential therapeutics across the GI tract, the
most common mode of transport is passive diffusion. An estimated 95% of
all marketed orally available drugs employ this method of transport.11a,b
Simply put, compounds that are absorbed via passive diffusion move
down a concentration gradient from areas ofL.high
Marco Lolli concentration,
University ofsuch
Torinoas
(UniTO)
the GI tract, to areas of low concentration, such as the systemic circula-
tion. Compound polarity can play a major role in passive diffusion, as inDiffusione paracellulare
1. Diffusione paracellulare
dipende dall’organizzazione cellulare (impossibile in
presenza di giunzioni serrate es. endotelio della
barriera ematoencefalica) e dalla dimensione del
farmaco (diametro del farmaco non deve superare
lo spazio tra le cellule)
Marco L. Lolli University of Torino (UniTO)Dimensioni complessive
Movimenti di rotazione di un soluto in soluzione attorno al proprio baricentro: sfera virtuale
diametro: ~ lunghezza della molecola
PAS
La dimensione complessiva (diametro) ed il PM sono correlati in modo grossolano
Acido acetilsalicilico Rifampicina
PM = 180 PM = 823
9Å 16 Å
Marco L. Lolli University of Torino (UniTO)Diffusione paracellulare
La diffusione paracellulare può iniziare a causa di un
gradiente di concentrazione nello strato cellulare
(diffusione passiva) o per un gradiente di pressione
idrostatica attraverso lo strato cellulare (filtrazione).
Se su una membrana cellulare viene esercitata una membrana
pressione idrostatica oppure esiste tra due lati un semipermeabile
gradiente osmotico, l’acqua e i soluti di piccole
dimensioni passeranno attraverso i pori della
membrana seguendo il gradiente pressorio.
diffusione libera
Nella maggior parte delle membrane i pori hanno un O
diametro di circa 7Å, attraverso I quali passano le H2N NH2
sostanze con peso < 100 Da, essenzialmente ioni e UREA
PM 60 Da
piccole molecole polari, come l’urea.
Marco L. Lolli University of Torino (UniTO)Diffusione paracellulare
Generalmente i canali tra le cellule endoteliali dei capillari sanguigni hanno un diametro di
circa 40 Å, sufficiente a consentire il passaggio a molecole con PM < 60 kDa, ma non in
tutti i compartimenti hanno lo stesso diametro.
Nel Sistema Nervoso Centrale (SNC) i capillari sono circondati da
uno strato di cellule gliali unite da tight junctions che circonda i
capillari e impedisce la filtrazione passiva; tale protezione va sotto il
nome di barriera emato-encefalica (BEE).
capillari continui
Nel glomerulo del rene ci sono i capillari fenestrati che hanno pori
di dimensioni maggiori, in modo da facilitare il passaggio dei soluti
di maggiori dimensioni (in minima parte anche piccole proteine,
come l’albumina) pertanto questa membrana è molto permeabile
capillari fenestrati e consente filtrazione di acqua e soluti.
Nel fegato ci sono i sinusoidi epatici che presentano aperture di
grandi dimensioni che consentono il passaggio anche
alle molecole proteiche in modo da consentire il
metabolismo di un’ampia gamma di composti chimici.
sinusoidi
Marco L. Lolli University of Torino (UniTO)Diffusione transcellulare
2. Diffusione transcellulare
un farmaco deve poter attraversare la membrana cellulare. Nel cammino dal sito di
somministrazione alla biofase il farmaco incontra diverse membrane cellulari, costituite da un
doppio strato di fosfolipidi in cui sono immerse delle proteine. Le proteine possono trovarsi
sia sul lato rivolto verso il liquido extracellulare sia su quello citoplasmatico. Dal punto di vista
del trasporto sono molto importanti le proteine transmembrana, che attraversano il bilayer
fosfolipidico. proteina
oligosaccaride glicoproteina periferica
glicolipide
proteina integrale
di membrana bilayer
bilayer
faccia esterna fosfolipidico
faccia citoplasmatica catene di
proteina acidi grassi fosfolipidi
proteina integrale proteine teste polari
idrofila
di membrana periferiche idrofile
Marco L. Lolli University of Torino (UniTO)Diffusione transcellulare
serina colina etanolamina inositolo I fosfolipidi sono esteri del glicerolo con due
NH3
+
N
+
NH3
+
molecole di acidi grassi a lunga catena.
COO -
H HO La presenza di insaturazioni, riduce il punto di
OH
O
O
P
O O O
HO OH fusione degli acidi grassi in quanto riduce le
O OH
H O interazioni di van der Waals e ha l’effetto di
Glicerolo fluidificare maggiormente il doppio strato
O
O
fosfolipidico; nelle membrane delle cellule
O O
• fosfatidilserina animali il colesterolo ha la stessa funzione.
• fosfatidilcolina Il terzo ossidrile alcolico è invece esterificato
• fosfatidiletanolamina con un aminoalcol (serina, colina, etanolamina)
acido oleico
• fosfatidilinositolo o un polialcol (inositolo), che in virtù della
presenza di cariche positive (testa ammonica
quaternaria) e negative (gruppi fosfato ionizzati)
conferisce un notevole grado di polarità,
facendo dei fosfolipidi molecole anfifiliche
(contengono sia un gruppo
idrofilo che idrofobo).
acido
palmitico
Marco L. Lolli University of Torino (UniTO)Diffusione transcellulare
ABSORPTION 255
Sebbene tutte le membrane cellulari abbiano una diversa permeabilità a
compound must be able to move through both the barriers of the gastroin-
seconda del tessuto, i principali meccanismi di passaggio attraverso le
testinal tract and the hepatocyte cell membrane in order to induce a thera-
membrane sono:
peutic response. Differences in permeability between various cell types can
also be an issue, as differences in membrane composition, junction tightness
A. Diffusione passiva
(the space between cells), and transporter protein activity can all influence
compound permeability. Compounds intended to modulate CNS func-
tions, for example, must traverse the blood–brain barrier, which is far more
B. Diffusione facilitata
restrictive in nature. Tight cellular junctions and higher levels of transport
protein expression designed to protect the brain from xenobiotics must be
C. Trasporto attivo
overcome in order for CNS targeted compounds to elicit a response.
The observed permeability of any given compound across a biologi-
cal barrier is the sum of five modes of transport: passive diffusion, active
D. Trasporto vescicolare
transport, endocytosis, paracellular transport, and efflux (Figure 6.10 ). In
(a) (b) (c) (d) (e)
FIGURE 6.10 (a) Passive diffusion; (b) Active transport; (c) Endocytosis; (d) Efflux; (e)
Marco L. Lolli University of Torino (UniTO)
Paracellular transport.Diffusione transcellulare
A. Diffusione passiva: processo secondo il quale le molecole
spontaneamente diffondono da una regione a più alta concentrazione
(fuori dalla cellula, OUT) ad una a più bassa concentrazione (dentro la
cellula, IN). E’ il principale meccanismo di passaggio attraverso le
membrane.
COut CIn
Anche molecole di grandi dimensioni o farmaci legati alle proteine non
possono diffondere attraverso le membrane. I farmaci liposolubili
attraversano le membrane lipidiche con facilità, mentre le molecole polari e
tutti i composti completamente ionizzati con velocità inferiore, o per nulla.
Marco L. Lolli University of Torino (UniTO)Diffusione transcellulare Diffusione di tipo convettivo Esistono dei canali acquosi nella membrana che permettono il passaggio di molecole idrofile non ioniche di piccole dimensioni (4-7Å, PM
Diffusione transcellulare
La velocità di diffusione passiva dipende dal gradiente di concentrazione
(Cout - Cin) del farmaco ai due lati della membrana e dal coefficiente di
ripartizione n-ottanolo/acqua (P) della sostanza, ed è direttamente
proporzionale all’area superficiale della membrana (A) e inversamente
proporzionale al suo spessore (h).
D ⋅ A ⋅ P ⋅ (C out − C in )
Legge di Fick: velocità di diffusione = -
h
D = coefficiente di diffusione
A = area superficiale della membrana
P = coefficiente di ripartrizione tra la membrana lipofila e la fase acquosa
Cout – Cin = differenza tra la conc. Esterna e la conc. Interna
H = spessore della membrana (75 Å)
Marco L. Lolli University of Torino (UniTO)B. Diffusione facilitata
- richiede trasportatore (carrier-proteina di membrana)
- non richiede energia
- è strutturalmente specifica
- segue la legge di Fick (gradiente di concentrazione) fino alla soglia di
saturazione
- possibilità di competizione per il carrier
Marco L. Lolli University of Torino (UniTO)Diffusione facilitata
Esempio di diffusione facilitata: glucosio (molto idrofilo) o vit. B12
Marco L. Lolli University of Torino (UniTO)C. Trasporto attivo
- Richiede un trasportatore
- È strutturalmente specifico
- Ha un limite di carico: è saturabile
- Fenomeni di competizione tra composti strutturalmente
simili possono ridurre l’assorbimento
Marco L. Lolli University of Torino (UniTO)Trasporto attivo
- Richiede energia*
- Si può verificare contro un gradiente di
concentrazione (non segue la legge di Fick)*
Es. amminoacidi, metildopa, penicillamina, levodopa, 5-
fluorouracile
NH2 NH2
N N
N N
O O O N N O O N N
O P
O
O P
O
O P
O
O O P
O
O P
O
O
+ PO3- + energia
O O
H H H H
ATP OH H
ADP OH H
(*) differenze dalla diffusione facilitata
Marco L. Lolli University of Torino (UniTO)D. Trasporto vescicolare
Formazione di una piccola cavità
nella membrana che gradualmente
circonda la molecola da trasportare
e la porta all’interno sottoforma di
vescicola (es. assorbimento del
vaccino di Sabin antipolio)
Endocitosi se il trasporto è verso
l’interno
Esocitosi se è verso l’esterno
Trancitosi se attraversa la cellula
Marco L. Lolli University of Torino (UniTO)Trasporto vescicolare
Caratteristico di particelle di dimensioni maggiori che non possono
penetrare nella cellula per trasporto passivo semplice o facilitato .
LDL
Internalizzazione
Riciclo dei Un esempio è costituito dall’endocitosi del
recettori in colesterolo, che essendo molto idrofobico e
membrana insolubile in acqua non può passare dall’intestino
(la frazione assunta con il cibo) o dal fegato (la
frazione sintetizzata) al sangue, disciolto nel
sangue o nel fluido extracellulare. Esso viene
invece veicolato da complessi lipoproteici (le
LDL), ossia sferette rivestite di un monostrato
fosfolipidico che ospitano al proprio interno il
separazione colesterolo.
recettori-LDL
LDL nel lisosoma
Marco L. Lolli University of Torino (UniTO)La Curva AUC
La velocità di assorbimento dipende dalla via di somministrazione, più corto è il tempo (tmax) richiesto per
raggiungere il livello di concentrazione massimo nel sangue (Cmax), più alta sarà la Cmax raggiunta e più
velocemente il livello nel plasma diminuirà. L’area sotto la curva (AUC) è indipendente dalla via di
somministrazione, a patto che la dose e la biodisponibilità siano uguali. La AUC può così essere usata per
determinare la biodisponibilità di un farmaco. Il rapporto dei valori di AUC determinati dopo
somministrazione orale o endovena per una data dose, corrisponde alla tendenza del farmaco di entrare
in circolo.
Marco L. Lolli University of Torino (UniTO)Curva AUC
L’assorbimento di un farmaco
dall’intestino causa l’aumento
della concentrazione del farmaco
nel sangue.
Poi il sangue lo trasporta nei vari
tessuti a seconda che questi siano
più o meno irrorati (il cervello è
quello tra i più irrorati).
Quindi passa dal fegato per
essere metabolizzato e dai reni
ed infine nelle urine per essere
escreto.
Marco L. Lolli University of Torino (UniTO)CARATTERISTICHE ANATOMO-FISIOLOGICHE DEGLI ORGANI
Ø permeabilità capillare
Ø grado di vascolarizzazione
Ø volume del tessuto
Permeabilità capillare:
Capillare continuo (cardiaco, scheletrico, cutaneo, connettivo, adiposo, polmonare)
Spazi intercellulari: 10 nm
Capillare fenestrato (glomeruli renali, ghiandole esocrine ed endocrine, mucosa
intestinale) Pori intracellulari: 50÷60 nm
Capillare discontinuo (fegato, milza e midollo osseo) Fessure intra ed
intercellulari (fino a 1 µm) permeabilità elevata: attraversati da grosse molecole
Marco L. Lolli University of Torino (UniTO)Metodologie di Sintesi e Sviluppo Farmaceutico
Synthesis and Development Pharmaceutical Methodologies
Laurea Magistrale in Chimica a.a. 2018/2019
L-ADME-T
Distribution
Marco L. Lolli University of Torino (UniTO)Assorbimento:
Passaggio del farmaco nel torrente circolatorio
q formulazione
q via di somministrazione (e.v.: l’assorbimento non è necessario)
q presenza di altri farmaci o alimenti
q proprietà chimico-fisiche del farmaco:
v solubilità
v grado di ionizzazione
v lipofilia
v dimensione molecolare
Marco L. Lolli University of Torino (UniTO)Permeability:
Capacità di superare le barriere biologiche
q proprietà chimico-fisiche del farmaco (lipofilia, caratteristiche acido-base, PM)
q caratteristiche anatomo-fisiologiche dell’organo
Marco L. Lolli University of Torino (UniTO)Distribuzione:
processo di trasferimento dal sangue ai tessuti
le concentrazioni tissutali del farmaco aumentano e la
sua concentrazione plasmatica diminuisce
q proprietà chimico-fisiche del farmaco (lipofilia, caratteristiche acido-base, PM)
q caratteristiche anatomo-fisiologiche dell’organo
q legame con le proteine plasmatiche
q tropismo tissutale
Marco L. Lolli University of Torino (UniTO)CARATTERISTICHE ANATOMO-FISIOLOGICHE DEGLI ORGANI
Ø permeabilità capillare
Ø grado di vascolarizzazione
Ø volume del tessuto
Permeabilità capillare:
Capillare continuo (cardiaco, scheletrico, cutaneo, connettivo, adiposo, polmonare)
Spazi intercellulari: 10 nm
Capillare fenestrato (glomeruli renali, ghiandole esocrine ed endocrine, mucosa
intestinale) Pori intracellulari: 50÷60 nm
diffusione paracellulare di farmaci non macromolecolari
Capillare discontinuo (fegato, milza e midollo osseo)
Fessure intra ed intercellulari (fino a 1 µm)
permeabilità elevata: attraversati da grosse molecole
Marco L. Lolli University of Torino (UniTO)Distribuzione a perfusione-limitata
Quando il flusso sanguigno è la tappa limitante la velocità di distribuzione del farmaco.
La permeabilità delle membrane tissutali è relazionata alle proprietà fisico-chimiche del
farmaco .
Per farmaci lipofili le membrane non rappresentano nessuna barriera e la distribuzione
dipende solo dalla velocità di perfusione nel tessuto.
Per questi farmaci vi è un rapido equilibrio tra concentrazione nel sangue e concentrazione
nei tessuti come polmoni, reni, fegato cuore e cervello, cioè quegli organi con un’alta
irrorazione sanguigna.
Un equilibrio più lento si trova nei muscoli, nelle ossa, nel tessuto adiposo, che ricevono un
volume di sangue per unità di massa decisamente inferiore.
Distribuzione a permeabilità-limitata
Quando la distribuzione del farmaco è limitata da un
lento passaggio attraverso le membrane nei tessuti.
Marco L. Lolli University of Torino (UniTO)Al contrario i farmaci
possono essere
Il passaggio del presenti nei tessuti a
farmaco dal concentrazioni più alte
sangue ai tessuti che nel plasma come
continua fino a conseguenza di un
quando non si gradiente di pH, ma
raggiunge un principalmente per
equilibrio tra la una più alta affinità per
forma diffusibile par ticolar i tipi di
del farmaco nei tessuti. Questo fatto è
tessuti e nel detto Accumulo.
s a n g u e , fi n o a
quando cioè la
concentrazione di
farmaco libero nel
plasma uguaglia Il farmaco può essere presente
quella nell’acqua ad alte concentrazioni nel
dei tessuti. plasma se è altamente legato
alle proteine plasmatiche.
Marco L. Lolli University of Torino (UniTO)Legame con le proteine plasmatiche
Molti sono i farmaci che si legano alle proteine
plasmatiche (albumina, β -globulina, α 1-
glicoproteina acida (AGA).
E’ importante conoscere la percentuale di farmaco
legato poichè il complesso farmaco-proteina è troppo
grosso e non può superare le barriere biologiche e
perciò ha una distribuzione limitata rimanendo
confinato nel plasma. Inoltre il complesso è
generalmente inattivo.
Un farmaco legato ad una proteina rimane più a
lungo nell’organismo, e la sua concentrazione libera, in
grado di agire, è decisamente più bassa rispetto alla
concentrazione totale.
Marco L. Lolli University of Torino (UniTO)Legame con le proteine plasmatiche
Questo equilibrio dipende dalla concentrazione dei
reagenti e dalla affinità del farmaco per la proteina.
Il legame F-P non è molto selettivo poichè una stessa proteina forma complessi con
numerosi farmaci strutturalmente diversi.
Ne consegue che diversi farmaci possono competere per gli stessi siti di legame di una
proteina plasmatica, provocandone a volte uno spiazzamento.
I farmaci lipofili, carichi o neutri, si legano molto, mentre farmaci idrofili sono poco legati
così come gli zwitterioni.
Il grado di legame con le proteine plasmatiche è alterato in alcune
condizioni fisiologiche (gravidanza, periodo neonetale) e patologiche
(insufficienza renale, e patica, stati infiammatori)
Marco L. Lolli University of Torino (UniTO)Legame con le proteine plasmatiche
Table 3. a) clogP calculated using Bio-Loom for Windows, vers.1.5; b) measured
using the shake flask-method. The “n.d.” notation indicates that the compound
was not tested in that specific assay.
Marco L. Lolli University of Torino (UniTO)Legame con le proteine plasmatiche
Table 4. Human serum stability and protein binding of compounds 2
and 4, as compared to brequinar.
Marco L. Lolli University of Torino (UniTO)Accumulo
I farmaci possono essere presenti nei tessuti a concentrazioni più alte che nel plasma
principalmente per una più alta affinità per particolari tipi di tessuti.
Es. farmaci lipofili, con alti LogP, si
accumulano nel tessuto adiposo.
Bisogna comunque ricordare che il
tessuto adiposo è pochissimo irrorato,
per questo l’accumulo è limitato ed
tiopentale morfina anche di conseguenza il rilascio dal
log P=10 log P=0.4 tessuto adiposo al sangue.
alto accumulo no accumulo
Pertanto l’accumulo nel tessuto adiposo risulta importante solo per quei farmaci lipofili
assunti cronicamente (es. Benzodiazepine).
Marco L. Lolli University of Torino (UniTO)I farmaci possono accumularsi nei tessuti
anche attraverso legami irreversibili con i
costituenti del tessuto stesso. Ad es. le
tetracicline, una classe di antibiotici, chelano il
Ca e per questo si accumulano nelle ossa e
nei denti.
Il tessuto in cui si accumulano certi farmaci è una potenziale riserva e a volte questo
accumulo può essere sfruttato per prolungare l’effetto del farmaco stesso.
Marco L. Lolli University of Torino (UniTO)Barriera ematoencefalica
I capillari nel cer vello sono costituiti da cellule
strettamente connesse che non contengono pori e
inoltre sono foderati di uno strato di grasso.
Perciò i farmaci per entrare nel cervello devono
capillari continui attraversare le membrane cellulari dei capillari e delle
cellule grasse che li rivestono.
La BEE permette di progettare farmaci idrofili che
agiranno selettivamente in altre parti del corpo senza
penetrare nel cervello.
D’altro canto i farmaci che devono agire nel cervello, devono attraversare la BEE e quindi
devono avere un numero minimo di gruppi polari, o averli mascherati
oppure devono essere trasportati all’interno con l’aiuto di carriers
o per pinocitosi (insulina).
Marco L. Lolli University of Torino (UniTO)CAPILLARI DELLA BARRIERA EMATOENCEFALICA (BEE o
BBB Blood Brain Barrier)
Ehrlich 1885 Goldmann 1913
Capillari della BBB:
q giunzioni serrate (tight junction): NON consentono la
diffusione paracellulare dei soluti
q rivestimento gliale
Diffusione passiva solo per farmaci
ü lipofili (logP0 = 2.3)
ü idrofili neutri di piccolissime dimensioni
(es. etanolo)
Marco L. Lolli University of Torino (UniTO)Barriera placentare
Separa il sangue materno dal sangue fetale: come possono
passare i nutrienti e i metaboliti, così anche i farmaci.
Farmaci lipofili la attraversano più facilmente.
Es. Alcol, nicotina, cocaina superano la barriera placentare
e passano nel circolo fetale, provocando effetti
imprevedibili sullo sviluppo del feto.
Farmaci ed altre tossine possono essere rimossa dal feto
dal sangue materno prima della nascita, ma dopo la nascita
rimangono nel feto che non ha le stesse capacità
metaboliche e di eliminazione della madre, così questi
farmaci avranno un tempo di emivita più lungo
e talvolta potranno avere effetti letali.
Marco L. Lolli University of Torino (UniTO)The Thalidomide accident
Thalidomide is not chi- rally stable in vivo. The (R)
isomer (left) is readily converted to the (S)
isomer (right). As a result, the pure (R) isomer is
no safer than the originally marketed racemic
material.
Thalidomide was brought to market in 1956 by Chemie Grünenthal GmbH
as an over the counter treatment for morning sickness in pregnant women.
By 1962, it had been withdrawn from markets across the globe as it had
been definitively linked to severe birth defect in the children of woman that
used it during their pregnancies.
Marco L. Lolli University of Torino (UniTO)Puoi anche leggere

















































