Manuale dell'impianto per il medico - Senza Senza II
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
Manuale dell’impianto per il medico Senza® Senza II® ONLY 2797 Inizio validità: settembre 2019

NEVRO CORP. Eventuali domande o dubbi sui prodotti Nevro Corp., inclusi eventuali eventi gravi verificatisi in relazione al dispositivo, dovranno essere inviati all’autorità competente locale e a: Nevro Corp. 1800 Bridge Parkway Redwood City, CA 94065, USA Tel: +1.650.251.0005 Fax: +1.650.251.9415 info@nevro.com Sponsor australiano MDSS GMBH Emergo Australia Schiffgraben 41 Level 20, Tower II, Darling Park D-30175 Hannover, 201 Sussex Street, Germania Sydney, NSW 2000 Australia © Copyright 2019, Nevro Corp. Tutti i diritti riservati. Nessuna parte della presente pubblicazione può essere riprodotta, trasmessa, trascritta, memorizzata in un sistema di archiviazione o tradotta in qualsivoglia lingua o linguaggio informatico, in qualsiasi forma o con qualsiasi mezzo, elettronico, magnetico, ottico, chimico, manuale o di altra natura, senza l’autorizzazione scritta di Nevro Corp. Marchi registrati: Nevro, Senza, Senza II, HF10 e il logo Nevro sono marchi di fabbrica di Nevro Corp. Marchio CE valido dal 4 maggio 2010 Nevro dichiara che il sistema Senza® è conforme ai requisiti essenziali e alle altre disposizioni pertinenti della direttiva sulle apparecchiature radio (2014/53/UE). IMPORTANTE: non sostituire o modificare i componenti del sistema di stimolazione del midollo spinale Senza o Senza II se l’operazione non è stata espressamente approvata da Nevro Corp. ATTENZIONE: la legge federale statunitense limita la vendita, la distribuzione e l’uso di questo dispositivo ai medici o su presentazione di prescrizione medica. 10186-ITA Rev O 2
Spiegazione dei simboli. Fare riferimento al prodotto per i simboli pertinenti. Simboli Descrizione SN Numero di serie LOT Codice lotto Data di fabbricazione Produttore Attenzione Numero di catalogo XX C XX C XXX F Limite di temperatura (conservazione) XX F STERILE EO Sterilizzato con ossido di etilene Data di scadenza Non utilizzare se la confezione è danneggiata Non riutilizzare Non risterilizzare Tenere all’asciutto ONLY Solo su prescrizione Consultare le istruzioni per l’uso in formato elettronico www.nevro.com Non sterile Radiazioni non ionizzanti Parte applicata di tipo B 10186-ITA Rev O 3
Parte applicata di tipo BF Non smaltire questo prodotto con i rifiuti urbani indifferenziati. Smaltire il prodotto secondo le disposizioni locali. A compatibilità RM condizionata Conditional Non compatibile con la RM Marchio CE di conformità 2797 Rappresentante autorizzato nella Comunità Europea 10186-ITA Rev O 4
Sommario 1. Descrizione del dispositivo ............................................................................................................. 6 2. Indicazioni per l’uso ....................................................................................................................... 8 3. Controindicazioni ........................................................................................................................... 8 4. Avvertenze..................................................................................................................................... 8 5. Precauzioni .................................................................................................................................. 12 6. Effetti avversi ............................................................................................................................... 16 7. Specifiche tecniche ....................................................................................................................... 18 a. Specifiche del sistema ......................................................................................................................... 18 b. Specifiche del caricabatterie ............................................................................................................... 18 c. Intervalli dei parametri di stimolazione .............................................................................................. 18 d. Qualità del servizio wireless................................................................................................................ 18 e. Sicurezza wireless................................................................................................................................ 18 f. Sicurezza informatica ........................................................................................................................... 19 g. Informazioni sulla telemetria .............................................................................................................. 19 h. Informazioni sulla ricarica wireless ..................................................................................................... 19 i. Interferenze elettromagnetiche........................................................................................................... 19 8. Identificazione del paziente .......................................................................................................... 22 9. Istruzioni per l’uso ........................................................................................................................ 23 10. Linee guida per l’impianto di prova ............................................................................................. 23 a. Prove temporanee e prove permanenti ......................................................................................... 23 b. Istruzioni preliminari ....................................................................................................................... 24 c. Posizionamento dell’elettrocatetere percutaneo........................................................................... 25 d. Posizionamento dell’elettrocatetere chirurgico ............................................................................. 25 e. Esecuzione di test intraoperatori .................................................................................................... 25 f. Preparazione della prova temporanea ........................................................................................... 26 g. Ancoraggio dell’elettrocatetere ...................................................................................................... 26 h. Collegamento di un’estensione all’elettrocatetere ........................................................................ 27 i. Tunnellizzazione percutanea dell’estensione ................................................................................. 28 j. Chiusura delle incisioni ................................................................................................................... 28 k. Collegamento dell’elettrocatetere o dell’estensione allo stimolatore di prova ............................. 29 11. Linee guida per l’impianto permanente ....................................................................................... 29 a. Istruzioni preliminari ....................................................................................................................... 29 b. Se il paziente ha già eseguito una prova temporanea .................................................................... 29 c. Se il paziente ha già eseguito una prova permanente .................................................................... 30 d. Impianto dell’IPG............................................................................................................................. 32 e. Espianto o sostituzione dell’IPG ...................................................................................................... 33 12. Informazioni sul dispositivo per il personale sanitario ................................................................. 33 a. Stimolatore di prova............................................................................................................................ 33 b. IPG ricaricabile .................................................................................................................................... 33 10186-ITA Rev O 5
1. Descrizione del dispositivo I sistemi di stimolazione del midollo spinale (SCS) Senza® e Senza II® sono dispositivi di neuromodulazione progettati per la stimolazione elettrica nella terapia del dolore cronico intrattabile del tronco e/o degli arti. I sistemi Senza e Senza II sono sistemi impiantabili e provvedono alla stimolazione tramite elettrocateteri impiantabili e un generatore di impulsi impiantabile (IPG) ricaricabile. L’IPG viene impiantato in una tasca sottocutanea ed è in grado di stimolare i nervi del midollo spinale se usato insieme a uno o più elettrocateteri. L’IPG viene controllato tramite il telecomando del paziente e/o il programmatore per il medico. Altri componenti dei sistemi Senza e Senza II includono uno stimolatore di prova esterno in grado di fornire la stessa stimolazione dell’IPG, estensioni per elettrocateteri, adattatori, caricabatterie e sistema di carica, cavi e accessori chirurgici. Di seguito sono riportati i dettagli dei sistemi Senza e Senza II: Dettagli del sistema Senza - Componenti principali Generatore di impulsi impiantabile (modelli Senza e Senza II): il generatore di impulsi impiantabile (IPG) è un dispositivo impiantabile ricaricabile dotato di 16 canali di uscita in grado di stimolare i nervi del midollo spinale tramite elettrocateteri. L’IPG è progettato per generare impulsi in uscita rettangolari ad accoppiamento capacitivo, bifasici, con bilanciamento di carica e regolazione di corrente. La testa dell’IPG contiene la bobina di ricarica e due porte che consentono l’inserimento degli elettrocateteri. La batteria ricaricabile è contenuta in un vano a chiusura ermetica posto all’interno dell’involucro ermetico in titano dell’IPG. Stimolatore di prova: lo stimolatore di prova è un piccolo dispositivo alimentato a batteria in grado di erogare la stessa stimolazione dell’IPG. Durante la fase di prova della stimolazione del midollo spinale, il soggetto indossa temporaneamente questo stimolatore di prova esterno per valutare l’efficacia della stimolazione prima dell'impianto del dispositivo permanente. Lo stimolatore di prova è collegato mediante cavi chirurgici agli elettrocateteri impiantati nel soggetto. Interfaccia tra IPG o stimolatore di prova e altri componenti Senza: il caricabatterie trasmette energia per via transcutanea per ricaricare la batteria dell’IPG. L’IPG e lo stimolatore di prova comunicano con il telecomando del paziente o il programmatore per il medico tramite l’unità di programmazione. I pazienti possono inoltre inviare comandi direttamente all’IPG o allo stimolatore di prova usando l’apposito telecomando. L’IPG è dotato anche di un interruttore magnetico che consente di interrompere la terapia tramite un magnete esterno. Telecomando del paziente: il telecomando del paziente è un piccolo dispositivo a batteria in grado di comunicare con l’IPG o lo stimolatore di prova. Include più comandi e indicatori per la comunicazione con questi componenti. Caricabatterie: il caricabatterie viene utilizzato dal paziente per ricaricare la batteria dell’IPG per via transcutanea. Si tratta di un dispositivo portatile alimentato con una batteria ricaricabile e può essere tenuto in una mano. Programmatore: il programmatore per il medico consente di programmare l’IPG o lo stimolatore di prova tramite l’unità di programmazione dotata di interfaccia utente grafica. Unità di programmazione: l’unità di programmazione è l’interfaccia del programmatore per il medico che consente la comunicazione con l’IPG o lo stimolatore di prova. 10186-ITA Rev O 6
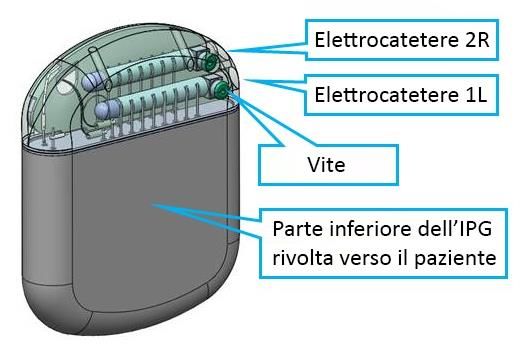
Elettrocateteri, estensioni e adattatori per elettrocateteri: sono disponibili due tipi di elettrocateteri, ovvero percutanei e chirurgici. Gli elettrocateteri Nevro sono progettati per essere usati con un IPG o uno stimolatore di prova per erogare la stimolazione. Gli elettrocateteri sono monouso e si interfacciano con l’IPG, le estensioni per gli elettrocateteri, il cavo chirurgico e gli accessori degli elettrocateteri. L’elettrocatetere percutaneo presenta un corpo isodiametrico in Pellethane 55D, con otto cavi a bassa impedenza. L’estremità del connettore prossimale presenta otto (8) contatti che si interfacciano con l’IPG Nevro e le estensioni degli elettrocateteri. L’estremità prossimale dell’elettrocatetere chirurgico ha due rami, ciascuno con 8 contatti, ed è identica all’estremità prossimale dell’elettrocatetere percutaneo. L’estremità distale dell’elettrocatetere è realizzata in silicone ed è dotata di 16 elettrodi distali. L’estremità prossimale dell’estensione dell’elettrocatetere è identica all’estremità prossimale dell’elettrocatetere percutaneo o chirurgico. L’estremità distale dell’estensione dell’elettrocatetere è progettata per accogliere l’estremità prossimale dell’elettrocatetere percutaneo o chirurgico. La struttura dell’estensione dell’elettrocatetere è identica per tutta la sua lunghezza a quella dell’elettrocatetere percutaneo. Gli adattatori M8 e S8 consentono al medico di collegare uno specifico elettrocatetere impiantato, rispettivamente Medtronic o St. Jude Medical, all’estensione dell’elettrocatetere Nevro o all’IPG. La struttura degli adattatori per elettrocateteri è identica a quella dell’estensione dell’elettrocatetere. Dettagli del sistema Senza - Accessori chirurgici Chiave dinamometrica: la chiave dinamometrica viene utilizzata per serrare le viti che bloccano l’elettrocatetere nell’IPG, per bloccare l’elettrocatetere in un’estensione/adattatore e per attivare il meccanismo di ritenzione sugli ancoraggi attivi. Ancoraggi degli elettrocateteri: gli ancoraggi degli elettrocateteri vengono utilizzati per ancorare l’elettrocatetere alla fascia o al legamento sopraspinoso. Ago di inserimento: l’ago di inserimento viene utilizzato durante la procedura di impianto per introdurre l’elettrocatetere percutaneo tra le vertebre nello spazio epidurale. Introduttore a spirale: l’introduttore a spirale viene utilizzato opzionalmente durante l’intervento chirurgico per creare un percorso per l’introduzione dell’elettrocatetere percutaneo nello spazio epidurale. Stiletti: gli stiletti servono per guidare l’elettrocatetere attraverso lo spazio epidurale fino alla posizione di impianto desiderata. Tappo per la porta dell’IPG: il tappo per la porta dell’IPG serve per sigillare la porta dell’IPG che non viene utilizzata quando si impianta un solo elettrocatetere. Cavi chirurgici: i cavi chirurgici consentono il collegamento elettrico e meccanico tra lo stimolatore di prova e gli elettrocateteri o le estensioni. 10186-ITA Rev O 7
Tunnellizzatore: il tunnellizzatore crea un tunnel sottocutaneo per gli elettrocateteri dal sito dell’IPG all’incisione mediana. Modello IPG: il modello IPG è un ausilio opzionale che assiste il medico nel corretto dimensionamento della tasca per l’impianto dell’IPG. Adattatore di prova Mx: l’adattatore di prova Mx serve per collegare un cavo chirurgico Medtronic allo stimolatore di prova Nevro. 2. Indicazioni per l’uso I sistemi di neuromodulazione Senza® e Senza II® sono previsti per la gestione del dolore cronico intrattabile del tronco e/o degli arti, compreso il dolore unilaterale o bilaterale associato a: Sindrome da fallimento chirurgico spinale Lombalgie refrattarie al trattamento medico Dolore dorsale Dolore agli arti inferiori Dolore agli arti superiori e al collo 3. Controindicazioni I sistemi Senza e Senza II non devono essere impiegati nei seguenti casi: • Pazienti non idonei all’intervento chirurgico. • Impossibilità di ottenere un efficace effetto analgesico durante la stimolazione di prova. • Pazienti incapaci di usare il sistema SCS. 4. Avvertenze Uso pediatrico - La sicurezza e l’efficacia della stimolazione del midollo spinale nei pazienti in età pediatrica non sono state verificate. Altri dispositivi impiantati attivi - I sistemi Senza e Senza II possono interferire con altri stimolatori impiantati, ad esempio pacemaker cardiaci e defibrillatori con funzioni di rilevazione, e possono provocare problemi di rilevazione e risposte anomale. Gli effetti sui sistemi Senza e Senza II di altri dispositivi impiantati (tra cui dispositivi per la stimolazione cerebrale profonda, stimolatori dei nervi periferici, pompe per il rilascio di farmaci e impianti cocleari) non sono noti. Sonno - I pazienti sottoposti a terapie che generano parestesie (sensazione di formicolio causata dalla stimolazione) possono scegliere di disattivare la stimolazione per evitare sensazioni sgradevoli durante il sonno. La terapia a 10 kHz non genera parestesie e pertanto la stimolazione può rimanere attiva durante il sonno. 10186-ITA Rev O 8
Uso di veicoli (ad esempio guida) o macchinari - I pazienti sottoposti a terapie che generano parestesie non devono azionare veicoli a motore, quali automobili o macchinari e apparecchiature potenzialmente pericolosi, quando la stimolazione è attiva. In questi casi è necessario disattivare prima la stimolazione. In questi pazienti, eventuali variazioni improvvise della stimolazione possono distogliere dal corretto utilizzo del veicolo, del macchinario o dell’apparecchiatura. La terapia a 10 kHz non genera parestesie ed è meno probabile che si verifichino variazioni improvvise della stimolazione che possono provocare distrazione quando la stimolazione è attiva e si usano veicoli in movimento, macchinari e apparecchiature. Sviluppo di calore durante la ricarica - Durante la ricarica, è possibile che la bobina di ricarica si scaldi. I pazienti possono avvertire disagio o calore elevato se il dispositivo viene ricaricato durante il sonno oppure se non viene usata la cintura del caricabatterie fornita in dotazione. Il caricabatterie non deve essere inoltre applicato su aree della cute prive di sensibilità. Diatermoterapia - Non impiegare la diatermia a onde corte, a microonde o a ultrasuoni su pazienti portatori del sistema di neuromodulazione. L’energia sviluppata dalla diatermia può essere trasmessa attraverso il sistema impiantato e danneggiare i tessuti in corrispondenza degli elettrodi, provocando gravi lesioni o la morte del paziente. Il sistema di neuromodulazione può subire danni, sia esso acceso o spento. Tomografia computerizzata (TC) - Prima della scansione tomografica, l’operatore deve acquisire scanogrammi per verificare se sono presenti dispositivi medici elettronici impiantati o esterni e, in caso affermativo, determinare la loro posizione rispetto all’area di scansione prevista. Per le procedure TC in cui il dispositivo medico si trovi all’interno o nelle immediate vicinanze dell’area di scansione prevista, l’operatore dovrà procedere come segue: • Determinare il tipo di dispositivo; • Se possibile, tentare di allontanare i dispositivi esterni dall’area di scansione; • Chiedere ai pazienti portatori di neurostimolatori di spegnere temporaneamente il dispositivo durante l’esecuzione della scansione; • Ridurre al minimo l’esposizione ai raggi X del dispositivo medico elettronico impiantato o esterno come segue: o Impiegare un’impostazione di corrente del tubo radiogeno più bassa possibile che consenta di ottenere la qualità delle immagini necessaria e o Accertarsi che il fascio di raggi X permanga sul dispositivo solo per pochi secondi. Nota importante: per le procedure TC che richiedono la scansione sopra il dispositivo medico per un tempo superiore a pochi secondi, come nel caso della perfusione TC o degli esami interventistici, il personale sanitario deve essere pronto ad adottare misure di emergenza per contrastare le eventuali reazioni avverse. Dopo aver eseguito la scansione TC direttamente sopra il dispositivo medico elettronico impiantato o esterno: • Chiedere al paziente di riaccendere il dispositivo se era stato spento prima della scansione. • Chiedere al paziente di verificare che il dispositivo funzioni correttamente, anche se era stato spento. • Raccomandare ai pazienti di contattare il proprio medico il prima possibile se sospettano che il dispositivo non funzioni correttamente dopo una scansione TC. 10186-ITA Rev O 9
Imaging a risonanza magnetica (RM) - I sistemi Senza e Senza II sono a compatibilità RM condizionata, vale a dire che la sicurezza è stata dimostrata solo in condizioni specifiche. L’esecuzione della scansione in condizioni diverse può provocare gravi lesioni al paziente o malfunzionamenti del dispositivo. Consultare le linee guida per la risonanza magnetica a 1,5 T e 3 T con i sistemi Senza sul sito Web di Nevro (www.nevro.com/physicianmanuals) per avvertenze e precauzioni relative all’esecuzione di indagini di risonanza magnetica su un paziente portatore del sistema Senza o Senza II. Dispositivi in ambienti ospedalieri/clinici - L’uso dei dispositivi o delle procedure mediche seguenti può danneggiare il sistema di stimolazione del midollo spinale (SCS) o disattivare la stimolazione. Dopo l’uso di questi dispositivi o procedure può essere necessario espiantare l’IPG a causa di danni permanenti. • Elettrocauterizzazione: non esporre l’IPG ad elettrocauterizzazione. Se è necessario effettuare l’elettrocauterizzazione con l’IPG impiantato, usare un elettrocauterio bipolare. Non usare elettrocauteri monopolari. • Defibrillazione esterna: la sicurezza della scarica di un defibrillatore esterno su pazienti portatori di sistema SCS non è stata accertata. • Litotrissia o ultrasuoni ad alta potenza: non impiegare questi dispositivi su pazienti con IPG impiantato. • Radioterapia: se si rendono necessarie applicazioni radioterapiche nei pressi dell’IPG, schermare l’area sopra l’IPG. • Ecografia: non eseguire sopra l’IPG. Se il paziente deve essere sottoposto a litotrissia, ultrasuoni ad alta potenza, elettrocauterizzazione, defibrillazione esterna, radioterapia o ecografia, osservare le seguenti precauzioni. • Spegnere l’IPG prima della procedura. • Usare i dispositivi alla massima distanza possibile dall’IPG. • Mantenere i campi, quali corrente, radiazioni o fasci di ultrasuoni ad alta potenza, a distanza dall’IPG. • I dispositivi devono essere impostati sul valore di energia più basso possibile. • Dopo la terapia o la procedura, verificare il corretto funzionamento dell’IPG aumentando gradualmente la stimolazione fino al livello desiderato. • Se sospetta che il dispositivo non funzioni correttamente dopo l’impiego di queste terapie o procedure, il paziente dovrà contattare il proprio medico. Interferenze elettromagnetiche (EMI) - L’energia elettromagnetica viene generata da apparecchiature presenti in ambienti domestici, professionali, clinici e pubblici. Le interferenze elettromagnetiche possono verificarsi quando l’energia è abbastanza elevata da interferire con la funzionalità del neurostimolatore. La maggior parte dei dispositivi elettrici e dei magneti di uso comune ha scarse probabilità di influire sul funzionamento del sistema SCS. Alcune apparecchiature possono tuttavia generare campi elettromagnetici potenti in grado di spegnere lo stimolatore (IPG o stimolatore di prova, TSM) o di provocare scosse o sobbalzi (vedere di seguito). I pazienti devono mantenersi sempre a distanza dalle aree con interferenze elettromagnetiche e spegnere lo stimolatore ogni volta che si trovano all’interno di tali aree. Di seguito sono elencati alcuni esempi di sorgenti che possono provocare potenti interferenze elettromagnetiche. 10186-ITA Rev O 10
• Rilevatori antitaccheggio o dispositivi di screening di sicurezza presso aeroporti, negozi o biblioteche. • Nota: è consigliabile che i pazienti richiedano assistenza per evitare di passare attraverso rilevatori antitaccheggio o dispositivi di screening di sicurezza. Se deve necessariamente passare attraverso un dispositivo di screening di sicurezza, il paziente dovrà spegnere lo stimolatore e riaccenderlo dopo aver attraversato il dispositivo di screening. • Linee elettriche e generatori • Saldatrici ad arco • Altoparlanti stereo magnetici di grandi dimensioni • Dispositivi di identificazione a radiofrequenza (RFID) Le interferenze elettromagnetiche potenti possono provocare quanto segue: Gravi lesioni nel paziente derivanti dal riscaldamento dei componenti impiantati del sistema di neurostimolazione e danno ai tessuti circostanti. Danni al sistema con la conseguente perdita o alterazione del controllo dei sintomi e la necessità di sostituzione chirurgica. Alterazioni funzionali del neurostimolatore, con conseguente accensione o spegnimento. Variazioni impreviste della stimolazione che provocano un aumento temporaneo della stimolazione o una stimolazione intermittente, percepita da alcuni pazienti come sobbalzi o scosse. Sebbene possano provocare disagio, le variazioni impreviste della stimolazione non danneggiano il dispositivo e non provocano lesioni nel paziente in modo diretto. In rari casi, le variazioni impreviste della stimolazione hanno provocato la caduta del paziente con conseguenti lesioni. I campi elettromagnetici potenti dovuti alla prossimità ad apparecchiature elettriche come telefoni cellulari e satellitari e sistemi radio possono interferire con la comunicazione radio tra telecomando e IPG. Un errore di comunicazione viene segnalato da tre bip. La comunicazione può essere ripristinata allontanandosi dall’apparecchiatura elettrica che provoca l’interferenza e ritentando l’operazione. Le scariche elettrostatiche (ESD) sono una comune fonte di interferenze elettromagnetiche e possono verificarsi quando una persona o un oggetto accumula una carica statica. Le scariche elettrostatiche peggiorano con la bassa umidità e i materiali sintetici. Se i terminali della batteria dello stimolatore di prova vengono esposti a scariche elettrostatiche, il dispositivo potrebbe reimpostarsi e arrestare la stimolazione. La stimolazione può essere riavviata seguendo le istruzioni riportate nella sezione “Come attivare la stimolazione” del Manuale del paziente. Per evitare l’arresto accidentale della stimolazione, non aprire il vano batterie mentre la stimolazione è attiva. Con le scariche elettrostatiche è possibile che il caricabatterie interrompa la ricarica dell’IPG. In questo caso è necessario riavviare la ricarica. Le scariche elettrostatiche possono essere ridotte al minimo tenendo il caricabatterie nell’apposito fodero durante la ricarica dell’IPG. L’unità di programmazione è idonea all'uso in aree industriali e in ambienti ospedalieri (secondo CISPR 11 Classe A). Se utilizzata in un ambiente residenziale (per il quale valgono di norma i requisiti di CISPR 11 Classe B), l’unità di programmazione potrebbe non fornire un’adeguata protezione ai servizi di comunicazione in radiofrequenza. Potrebbe essere necessario adottare misure correttive, ad esempio riorientare o riposizionare l’unità di programmazione. Nota: CISPR-Comitato Internazionale Speciale per le Interferenze Radio 10186-ITA Rev O 11
Il telecomando, il caricabatterie e lo stimolatore di prova sono idonei all'uso in un ambiente residenziale (secondo CISPR 11 Classe B). AVVERTENZA: evitare di collocare il sistema Senza adiacente o sovrapposto ad altre apparecchiature, in quanto ciò potrebbe causare malfunzionamenti. Se l’uso in tale configurazione si rende necessario, tenere sotto osservazione il sistema Senza e le altre apparecchiature per verificarne il corretto funzionamento. AVVERTENZA: l’uso di accessori diversi da quelli specificati o forniti da Nevro può comportare un aumento delle emissioni elettromagnetiche o una minore immunità elettromagnetica del sistema Senza, con conseguenti malfunzionamenti. AVVERTENZA: le apparecchiature di comunicazione portatili a radiofrequenza (incluse le periferiche come i cavi delle antenne e le antenne esterne) devono essere utilizzate a una distanza minima di 15 cm da qualsiasi componente del sistema Senza, inclusi i cavi. In caso contrario, il sistema Senza potrebbe non funzionare correttamente. Ablazione a radiofrequenza o a microonde - La sicurezza dell’ablazione a radiofrequenza o a microonde nei pazienti con sistema di neuromodulazione impiantato non è stata determinata. Le correnti elettriche indotte possono provocare riscaldamento, soprattutto in corrispondenza degli elettrodi dell’elettrocatetere, con conseguenti danni ai tessuti. 5. Precauzioni Pazienti non idonei - Non impiantare un sistema di stimolazione del midollo spinale (SCS) se un paziente non è ritenuto idoneo per l’intervento chirurgico. L’impianto di un sistema SCS comporta rischi simili a quelli degli interventi di chirurgia spinale, tra cui fuoriuscita di liquido spinale, cefalea, edema, ematomi, emorragie, infezioni o paralisi. Gravidanza - La sicurezza e l’efficacia della stimolazione del midollo spinale non sono state verificate durante la gravidanza o l’allattamento. Attività del paziente - I pazienti che usano terapie che generano parestesie possono manifestare un aumento delle parestesie durante i cambiamenti posturali o i movimenti improvvisi. Tali pazienti dovranno ridurre l’ampiezza o disattivare la stimolazione prima di eseguire cambiamenti posturali, quali movimenti di stiramento o sollevamento delle braccia sopra la testa. In presenza di sensazioni sgradevoli, è necessario spegnere l’IPG. La stimolazione a 10 kHz non genera parestesie, pertanto i pazienti non avvertiranno sensazioni sgradevoli con i cambiamenti posturali o i movimenti. Non sarà pertanto necessario modificare l’ampiezza dei programmi in caso di cambiamenti posturali o movimenti. Attività del paziente e dislocazione degli elettrocateteri - Raccomandare al paziente di non effettuare movimenti repentini e accentuati di flessione, estensione o rotazione, in particolare nelle prime settimane successive all’intervento. Durante tali movimenti, un elettrocatetere può spostarsi dalla posizione originale, compromettendo l’erogazione della terapia, il paziente potrebbe avere bisogno della riprogrammazione dell'impianto o potrebbe essere necessario riposizionare l'elettrodo con un altro intervento. 10186-ITA Rev O 12
Immersioni subacquee e camere iperbariche - I pazienti devono evitare le immersioni subacquee a profondità superiori a 35 metri e le camere iperbariche a pressioni superiori a 4,5 ATM. La pressione presente a oltre 35 metri di profondità o 4,5 ATM può danneggiare i sistemi Senza e Senza II. Conservazione - I componenti e gli accessori del sistema devono essere conservati alle temperature consentite. Temperature eccessivamente calde o fredde potrebbero danneggiare i componenti; particolarmente dannoso potrebbe rivelarsi l’eccessivo calore. I dispositivi vanno conservati in aree a temperatura regolata entro i limiti di temperatura accettabili. Non esporre i componenti a liquidi o eccessiva umidità. • La temperatura di conservazione dell’IPG, dell’elettrocatetere, dell’estensione per elettrocatetere e del caricabatterie deve essere compresa nell’intervallo 0 °C - 45 °C (32 °F - 113 °F). • La temperatura di conservazione dello stimolatore di prova e del telecomando del paziente deve essere compresa tra -20 e 60 °C (tra -4 e 140 °F). Sterilizzazione - Questo dispositivo è esclusivamente monouso e non deve essere risterilizzato. • Prima di aprire la confezione sterile, ispezionare l’indicatore di sterilizzazione e la confezione stessa. • Non utilizzare il contenuto della confezione se questa appare rotta o lacerata oppure se si sospetta una contaminazione a causa di un sigillo non integro della confezione sterile. • Non utilizzare eventuali componenti che mostrino segni di danno. • Non risterilizzare la confezione o il contenuto, dato il rischio di infezioni e di malfunzionamenti del dispositivo. • Non utilizzare dopo la data di scadenza. • Tutti i componenti impiantati sono esclusivamente monouso. Non riutilizzarli. Manipolazione - I componenti e gli accessori del sistema devono essere maneggiati con cura. Non lasciarli cadere e non immergerli in acqua. Evitare inoltre di urtare i componenti del sistema contro superfici dure e maneggiarli sempre con estrema attenzione. Sebbene siano stati eseguiti test di affidabilità volti a garantire la qualità della fabbricazione e delle prestazioni, lasciar cadere i dispositivi su superfici dure, immergerli in acqua o maneggiarli con disattenzione può provocare danni permanenti a componenti e accessori. Non collegare il caricabatterie a prese di corrente nei pressi dell’acqua. Manipolazione degli elettrocateteri e delle estensioni - Maneggiare elettrocateteri ed estensioni seguendo queste linee guida: • L’elettrocatetere e l’estensione devono essere sempre maneggiati con cura. • Non piegare bruscamente l’elettrocatetere o l’estensione. • Non piegare, schiacciare o tendere eccessivamente l’elettrocatetere o l’estensione. • Non applicare una coppia di serraggio eccessiva (torsione) all’elettrocatetere o all’estensione. Non applicare suture direttamente sull’elettrocatetere o sull’estensione. • Per applicare una sutura attorno all’elettrocatetere, utilizzare gli ancoraggi forniti in dotazione. • Non forzare l’inserimento dell’elettrocatetere nello spazio epidurale. Utilizzare l’introduttore prima di introdurre l’elettrocatetere. • Creare un’asola per ridurre la tensione sull’elettrocatetere. • Non sottoporre l’elettrocatetere a una tensione eccessiva. • Non utilizzare strumenti appuntiti per maneggiare l’elettrocatetere o l’estensione. • Eliminare ogni traccia di fluidi corporei (ad esempio sangue) dall’estremità prossimale dell’elettrocatetere prima di collegarlo a qualsiasi altro componente. • Eliminare ogni traccia di fluidi corporei (ad esempio sangue) dallo stiletto prima di introdurlo o reintrodurlo nell’elettrocatetere. • Non esercitare forza eccessiva quando si inserisce lo stiletto nell’elettrocatetere. 10186-ITA Rev O 13
Manipolazione dell’IPG - Seguire queste linee guida quando si maneggia l’IPG: • Evitare di maneggiare l’IPG in modo brusco. • Fare attenzione a non far cadere l’IPG. Se il dispositivo è caduto su una superficie dura, non utilizzarlo e restituirlo a Nevro Corp. Compatibilità del sistema - Non utilizzare cavi o adattatori non espressamente autorizzati da Nevro Corp. Stimolazione magnetica transcranica (TMS) e terapia elettroconvulsivante (TEC) - La sicurezza della TMS o della TEC non è stata valutata in pazienti con sistema di neuromodulazione impiantato. Le correnti elettriche indotte possono provocare riscaldamento, soprattutto in corrispondenza degli elettrodi dell’elettrocatetere, con conseguenti danni ai tessuti. Stimolazione elettrica transcutanea dei nervi - Non applicare elettrodi per la stimolazione elettrica transcutanea dei nervi (TENS) in una modalità che provochi il passaggio della corrente della TENS sopra parti del sistema di neurostimolazione. Se ritengono che la TENS possa interferire con il neurostimolatore impiantato, i pazienti dovranno interrompere l’uso della TENS fino a quando non si consulteranno con il proprio medico. Dolore post-operatorio - Nei giorni successivi all’intervento, il paziente potrà avvertire dolore nella sede dell’impianto. Si tratta di un fenomeno tipico negli interventi di impianto di stimolatori del midollo spinale. Posizione dell’IPG e manipolazione da parte del paziente - Raccomandare ai pazienti di non torcere o ruotare l’IPG. Se l’IPG impiantato si capovolge, il caricabatterie può non essere in grado di ricaricare l’IPG. La manipolazione dell’IPG da parte del paziente può provocare il progressivo assottigliamento della cute soprastante. Infezioni - Adottare procedure adeguate di controllo delle infezioni. Se il paziente avverte una sensazione di disagio persistente o osserva un eccessivo arrossamento della cute attorno all’incisione, occorrerà escludere la presenza di infezioni. Le infezioni correlate allo stimolatore del midollo spinale (SCS) possono rendere necessario l’espianto dei componenti. Non utilizzare il caricabatterie se l’incisione non appare sufficientemente guarita. Il caricabatterie e la cintura del caricabatterie non sono sterili e non devono venire a contatto con l’incisione. Temperatura di esercizio - La temperatura di esercizio del telecomando del paziente è compresa tra 10 e 40 °C (tra 50 e 104 °F). La temperatura di esercizio dello stimolatore di prova è compresa tra 10 e 38 °C (tra 50 e 100 °F). Quando il caricabatterie è collegato alla presa di corrente per la ricarica, la temperatura di esercizio del sistema di ricarica è compresa tra 10 e 40 °C (tra 50 e 104 °F). Quando il caricabatterie carica l’IPG, la temperatura di esercizio del sistema di ricarica è compresa tra 10 e 30 °C (tra 50 e 86 °F). Pulizia dello stimolatore di prova, del telecomando e del sistema di ricarica - Questi componenti possono essere puliti strofinandone tutte le superfici con un panno morbido inumidito con acqua, alcool isopropilico o detergente neutro, senza esercitare eccessiva pressione. Eventuali residui possono essere asportati con un panno asciutto. Non utilizzare detergenti abrasivi. Impedire la penetrazione di umidità all’interno dei componenti. Telefoni cellulari - Allo stato attuale non è noto l’effetto dei telefoni cellulari sul sistema di neuromodulazione. 10186-ITA Rev O 14
Guasto dell’IPG - Se l’IPG del paziente non effettua la stimolazione neanche dopo la sua ricarica completa o la sostituzione delle batterie del telecomando, spegnere l’IPG e rivolgersi a Nevro Corp. Quando la frequenza delle ricariche diventa poco pratica per il paziente, può essere necessario sostituire l’IPG. Rivolgersi a Nevro Corp. Smaltimento del dispositivo - Non smaltire l’IPG, il telecomando del paziente o il caricabatterie gettandoli nel fuoco. A contatto con le fiamme, la batteria di questi dispositivi può esplodere. In caso di cremazione del paziente, l’IPG deve essere espiantato. Tutti gli IPG espiantati devono essere restituiti a Nevro Corp. Non smaltire alcun componente elettrico, batterie incluse, con i rifiuti destinati alla raccolta indifferenziata. Tutti i componenti elettrici, batterie incluse, devono essere smaltiti conformemente alle normative locali. Smaltire gli accessori chirurgici nell’apposito contenitore per rifiuti biologici pericolosi, in conformità alle normative locali. Dichiarazioni FCC: IPG Senza, stimolatore di prova e telecomando del paziente - ID FCC dell’IPG Senza: XKYIPG1000, XKYIPG1500, XKYIPG2000 ID FCC dello stimolatore di prova: XKYEXTS1000 ID FCC del telecomando del paziente: XKYPTRD1000 Nota: • Questo dispositivo è stato testato e ritenuto conforme ai limiti fissati per i dispositivi digitali di classe B, conformemente alla Parte 15 del regolamento FCC. Questi limiti sono volti a fornire una ragionevole protezione dalle interferenze dannose in un’installazione residenziale. Questo dispositivo genera, utilizza e può irradiare energia in radiofrequenza e, se non installato e utilizzato conformemente alle istruzioni, può provocare interferenze nocive con le comunicazioni radio. Non si garantisce tuttavia che non si verifichino interferenze in una specifica installazione. Se questo dispositivo provoca interferenze con la ricezione radio o televisiva, individuabili spegnendolo e riaccendendolo, si consiglia di porvi rimedio ricorrendo a una o più delle seguenti misure: o Riorientare l’antenna ricevente. o Aumentare la distanza tra il dispositivo e l’antenna. o Collegare il dispositivo a una presa su un circuito diverso da quello a cui è collegato il ricevitore. o Rivolgersi al rivenditore o a un tecnico radio/TV esperto per richiedere assistenza. • Questo dispositivo non deve interferire con stazioni che operano nella banda di frequenza 400,150 – 406,000 MHz per ausili meteorologici, satelliti meteorologici e servizi satellitari di esplorazione terrestre e deve accettare eventuali interferenze ricevute, incluse quelle che possono influire negativamente sul funzionamento. • Non sostituire o modificare i componenti del sistema di stimolazione del midollo spinale Senza o Senza II se l’operazione non è stata espressamente approvata da Nevro Corp. Dichiarazioni FCC: Unità di programmazione - ID FCC dell’unità di programmazione: XKYWAND1000, XKYWAND1001 Nota: • Questo dispositivo è stato testato e ritenuto conforme ai limiti fissati per i dispositivi digitali di classe A, conformemente alla Parte 15 del regolamento FCC. Questi limiti sono volti a fornire una ragionevole protezione dalle interferenze dannose quando il dispositivo viene utilizzato in un ambiente commerciale. Questo dispositivo genera, utilizza e può irradiare energia in radiofrequenza e può provocare interferenze nocive con le comunicazioni radio se non installato e utilizzato conformemente al manuale di istruzioni. Il funzionamento di questo dispositivo in un’area residenziale può provocare interferenze nocive. In tal caso, l’operatore dovrà eliminare le interferenze a proprie spese. 10186-ITA Rev O 15
• Questo dispositivo non deve interferire con stazioni che operano nella banda di frequenza 400,150 – 406,000 MHz per ausili meteorologici, satelliti meteorologici e servizi satellitari di esplorazione terrestre e deve accettare eventuali interferenze ricevute, incluse quelle che possono influire negativamente sul funzionamento. • Non sostituire o modificare i componenti del sistema di stimolazione del midollo spinale Senza o Senza II se l’operazione non è stata espressamente approvata da Nevro Corp. 6. Effetti avversi L’uso di un sistema SCS è associato a potenziali rischi che possono essere correlati alla procedura di impianto, alla stimolazione o a un componente del sistema. Raccomandare al paziente di contattare il proprio medico in caso di eventi avversi correlati al dispositivo. I possibili rischi includono quanto segue: Procedura di impianto e rischi medici Rischi associati all’anestesia, incluso l’arresto cardiaco Complicanze chirurgiche, ad esempio infezioni, cellulite, ascesso, febbre, sepsi, sanguinamento Fuoriuscita di liquido cerebrospinale Ipotensione intracranica Ematoma, sieroma o trombosi Emorragia epidurale Guarigione insufficiente o inadeguata dell’incisione, deiscenza della ferita Sensibilità o dolore temporaneo o persistente nel sito dell’impianto Migrazione dell’elettrocatetere, con conseguente controllo inefficace del dolore o altre variazioni indesiderate della stimolazione Posizionamento non ottimale o migrazione dell’elettrocatetere o dell’IPG che richiede la revisione o l’espianto Compressione del midollo spinale; lesioni a carico di nervi, radici dei nervi o midollo spinale Paralisi Morte Stimolazione Perdita dell’effetto analgesico, perdita di parestesia, parestesia sgradevole Incremento del dolore Stimolazione indesiderata provocata da cambiamenti cellulari che si verificano con il tempo nel tessuto attorno agli elettrodi, dalla migrazione degli elettrodi, dall’allentamento dei collegamenti elettrici o da guasti degli elettrocateteri Stimolazione sgradevole del tessuto attorno agli elettrocateteri, inclusi la pelle e i muscoli Altre sensazioni sgradevoli come formicolio o pizzicore Debolezza, disturbi della coordinazione o intorpidimento Componenti impiantati del dispositivo Reazione tissutale o allergia ai materiali impiantati Dolore persistente nel sito dell’impianto (elettrocatetere o IPG) Guasto di componenti del dispositivo o della batteria, compresa la rottura o lo spostamento (migrazione) dell’elettrocatetere, malfunzionamenti dell'hardware, collegamenti lenti, cortocircuiti o circuiti aperti e usura dell'isolamento dell’elettrocatetere 10186-ITA Rev O 16
Guasto o malfunzionamento con conseguente controllo inefficace del dolore o altre variazioni indesiderate della stimolazione e che può richiedere l'espianto e il reimpianto Erosione cutanea o sieroma nel sito dell’elettrocatetere o dell’IPG Ulcere da pressione Sorgenti esterne di interferenze elettromagnetiche che causano il malfunzionamento del dispositivo e possono compromettere la stimolazione L’esposizione alla risonanza magnetica (RM) può provocare il riscaldamento del tessuto, artefatti di immagine, tensioni indotte nell’IPG e/o negli elettrocateteri e dislocazione degli elettrocateteri Infezioni Formazione di una massa epidurale nel sito dell’elettrocatetere: sebbene l’incidenza sia rara (14 casi in 30 anni; vedere il riferimento di seguito), nel corso di mesi o anni l’impianto permanente di un elettrocatetere a piattina o di un elettrocatetere percutaneo per SCS può provocare la formazione di una massa epidurale attorno all’elettrocatetere, con conseguente compressione del midollo spinale. L’effetto della compressione midollare può variare dalla debolezza muscolare alla quadriplegia progressiva. Se un paziente con un elettrocatetere per SCS presenta un nuovo deficit neurologico, è necessario prendere in considerazione come potenziale causa la compressione del midollo spinale dovuta alla formazione di una massa tissutale reattiva. Se viene identificata una massa epidurale in un paziente asintomatico, è opportuno porre in atto un monitoraggio periodico. (professional.medtronic.com/wcm/groups/mdtcom_sg/@mdt/@neuro/documents/documents /scs-compression-ltr-feb2014.pdf) Componenti esterni del dispositivo Reazione tissutale o allergia a materiali esterni Effetti sgradevoli da riscaldamento, fastidio o bruciore/ustioni 10186-ITA Rev O 17
7. Specifiche tecniche a. Specifiche del sistema Parametro Intervallo Frequenza 2 - 10.000 Hz Durata dell’impulso 20 μs - 1 ms Ampiezza 0 - 15 mA b. Specifiche del caricabatterie La tabella seguente elenca le specifiche tecniche del caricabatterie. Ingresso c.a. del caricabatterie: Parametro Specifica Frequenza Da 50 a 60 Hz Tensione Da 100 a 240 V c.a. Corrente di ingresso 0,2 A max Sono disponibili ulteriori informazioni tecniche, comprese le indicazioni e dichiarazioni del produttore su emissioni e immunità elettromagnetiche. Per richiedere queste informazioni rivolgersi a Nevro Corp. c. Intervalli dei parametri di stimolazione La tabella seguente elenca l’impedenza massima a cui può essere erogata la corrente massima di 15 mA alla durata massima dell’impulso (1 ms, a una frequenza massima di 400 Hz) e alla frequenza massima (10.000 Hz, a una durata massima dell’impulso di 30 µs). Frequenza Ampiezza Durata massima Impedenza massima dell’impulso massima 400 Hz 15 mA 1 ms 1.270 Ω 10.000 Hz 15 mA 30 µs 1.080 Ω d. Qualità del servizio wireless I sistemi Senza e Senza II impiegano un sistema di comunicazione wireless nella banda di frequenza MedRadio (402 - 405 MHz). Questa banda è riservata ai dispositivi medici impiantabili. La portata tipica della comunicazione è inferiore a 1,5 metri (5 piedi) tra il telecomando del paziente/l’unità di programmazione e il generatore di impulsi impiantabile (IPG)/lo stimolatore di prova. Prima di ogni comunicazione, il telecomando del paziente/l’unità di programmazione esegue la scansione di 8 canali nella banda e seleziona il canale con meno interferenze per la comunicazione. Viene verificata l’accuratezza di tutte le comunicazioni. Qualsiasi comunicazione contenente errori irrisolvibili viene respinta e la comunicazione viene ritentata automaticamente. Se i tentativi non hanno esito positivo, l’utente viene informato dell’errore di comunicazione. e. Sicurezza wireless I sistemi Senza e Senza II hanno una portata di telemetria inferiore a 1,5 metri (5 piedi). Il telecomando del paziente è associato in modo univoco a un generatore di impulsi impiantabile (IPG) o a uno stimolatore di prova specifico e può comunicare esclusivamente con tale dispositivo. Il generatore di impulsi impiantabile e lo stimolatore di prova non rispondono a comunicazioni che non provengano da un dispositivo collegato (ovvero associato all’IPG). L’integrità dei dati comunicati viene garantita da ulteriori meccanismi. 10186-ITA Rev O 18
f. Sicurezza informatica I sistemi operativi e le applicazioni software vengono gestiti e aggiornati per proteggerli dalle lacune di sicurezza identificate per i componenti del sistema Nevro collegati in rete. Nevro assicura che i dati contenuti in tali sistemi/applicazioni siano adeguatamente protetti/crittografati, conformemente alle linee guida normative e agli standard di settore applicabili. g. Informazioni sulla telemetria I sistemi Senza e Senza II impiegano un sistema di comunicazione wireless nella banda di frequenza MedRadio (402 - 405 MHz). Il sistema di comunicazione wireless implementa la modulazione Frequency Shift Keying (FSK). La larghezza di banda di ciascuno degli 8 canali di frequenza non supera i 300 kHz e la potenza irradiata isotropica efficace del trasmettitore (EIRP) non supera i -16 dBm (25 µW). Consultare il Manuale del paziente per altre informazioni sull’ottimizzazione della comunicazione. Per determinare le distanze raccomandate tra il sistema Senza o Senza II e altri trasmettitori, consultare le tabelle nella sezione “Interferenze elettromagnetiche”. h. Informazioni sulla ricarica wireless I sistemi Senza e Senza II utilizzano una frequenza di ricarica di 410 - 485 kHz. La distanza di ricarica tra il caricabatterie e l’IPG è compresa tra 0 e 2,5 cm. Per ottimizzare la ricarica, consultare le istruzioni riportate nel Manuale del paziente. i. Interferenze elettromagnetiche Indicazioni e dichiarazione del produttore - Emissioni elettromagnetiche I sistemi Senza e Senza II sono progettati per l’uso nell’ambiente elettromagnetico specificato di seguito. È responsabilità dell’acquirente o dell’operatore del sistema Senza o Senza II garantirne l’uso in tale ambiente. Test sulle emissioni Conformità Indicazioni per l’ambiente elettromagnetico Emissioni RF CISPR 11 Gruppo 1 I sistemi Senza e Senza II impiegano l’energia RF solo per le funzioni interne. Le emissioni RF sono pertanto molto basse ed è improbabile che provochino interferenze nelle apparecchiature elettroniche che si trovano nelle vicinanze. Emissioni RF CISPR 11 Classe B I sistemi Senza e Senza II sono adatti all’uso in tutti gli Classe A (unità di ambienti, compresi quelli residenziali e quelli direttamente programmazione) collegati alla rete elettrica pubblica a bassa tensione usata per Emissioni armoniche Classe B gli edifici adibiti a uso domestico. IEC 61000-3-2 Fluttuazioni di tensione/ Conforme L’unità di programmazione è idonea per l’uso in tutti gli Emissioni flicker ambienti, eccetto gli ambienti domestici e quelli collegati IEC 61000-3-3 direttamente alla rete elettrica pubblica a bassa tensione che alimenta gli edifici usati per scopi domestici. 10186-ITA Rev O 19
Guida e dichiarazione del produttore - Immunità elettromagnetica I sistemi Senza e Senza II sono progettati per l’uso nell’ambiente elettromagnetico specificato di seguito. È responsabilità dell’acquirente o dell’operatore del sistema Senza/Senza II garantirne l’uso in tale ambiente. Test di immunità Livello di test Livello di Indicazioni per l'ambiente elettromagnetico IEC 60601 conformità Scariche ± 2/4/8/15 kV ± 2/4/8/15 kV I pavimenti devono essere di legno, cemento elettrostatiche in aria in aria o in piastrelle di ceramica. Se i pavimenti (ESD) IEC 61000-4-2 ± 8 kV a contatto ± 8 kV a contatto sono coperti con materiali sintetici, l’umidità relativa non deve essere inferiore al 30%. Transitori elettrici ± 2 kV, PRF 100 kHz ± 2 kV, PRF 100 kHz La qualità della rete elettrica deve essere veloci/burst quella di un tipico ambiente commerciale IEC 61000-4-4 o ospedaliero. Sovratensione ± 1 kV da linea ± 1 kV da linea La qualità della rete elettrica deve essere IEC 61000-4-5 a linea a linea quella di un tipico ambiente commerciale ± 2 kV da linea ± 2 kV da linea o ospedaliero. a terra a terra Cali di tensione, 0% UT per 0,5 cicli, 0% UT per 0,5 cicli, La qualità della rete elettrica deve essere brevi interruzioni 0˚, 45˚, 90˚, 135˚, 0˚, 45˚, 90˚, 135˚, quella di un tipico ambiente commerciale e variazioni 180˚, 225˚, 270˚ 180˚, 225˚, 270˚ o ospedaliero. Se l’utente del sistema Senza/ di tensione e 315˚ e 315˚ Senza II richiede un funzionamento continuo sulle linee di durante le interruzioni dell’alimentazione di alimentazione 0% UT 0% UT rete, si raccomanda di alimentare il sistema in ingresso per 1 ciclo per 1 ciclo Senza/Senza II mediante un gruppo di IEC 61000-4-11 continuità o una batteria. 70% UT per 25 cicli 70% UT per 25 cicli 0% UT per 250 cicli 0% UT per 250 cicli Campo magnetico 30 A/m 30 A/m I campi magnetici a frequenza di rete devono a frequenza di rete attestarsi ai livelli caratteristici di una tipica (50/60 Hz) ubicazione in un tipico ambiente commerciale IEC 61000-4-8 o ospedaliero. NOTA - UT indica la tensione di rete c.a. prima dell’applicazione del livello di test. 10186-ITA Rev O 20
Puoi anche leggere

















































