LA SCLEROSI MULTIPLA È PROGRESSIVA SIN DALL'INIZIO1
←
→
Trascrizione del contenuto della pagina
Se il tuo browser non visualizza correttamente la pagina, ti preghiamo di leggere il contenuto della pagina quaggiù
LA SCLEROSI MULTIPLA
È PROGRESSIVA SIN DALL’INIZIO1
La letteratura suggerisce che il trattamento
con terapie immunologiche ad elevata efficacia
risulta più potente nel sopprimere le ricadute
se iniziato precocemente dalla diagnosi di SM.2
LA PRIMA E UNICA TERAPIA3,4 APPROVATA
PER SOPPRIMERE LA PROGRESSIONE
DELLA SMR E DELLA SMPP5*
Medicinale sottoposto a monitoraggio addizionale. Ciò permetterà la rapida
identificazione di nuove informazioni sulla sicurezza. Agli operatori sanitari è
richiesto di segnalare qualsiasi reazione avversa sospetta. Vedere paragrafo 4.8 per
informazioni sulle modalità di segnalazione delle reazioni avverse.5
*OCREVUS® è indicato per il trattamento di pazienti adulti affetti da:5
• forme recidivanti di sclerosi multipla (SMR) con malattia attiva definita in base
alle caratteristiche cliniche o radiologiche (vedere paragrafo 5.1).
• sclerosi multipla primariamente progressiva (SMPP) in fase precoce in termini
di durata della malattia e livello di disabilità, e con caratteristiche radiologiche tipiche
di attività infiammatoria (vedere paragrafo 5.1).
Tasso annualizzato di recidiva
I pazienti trattati in modo continuativo con OCREVUS hanno mantenuto
bassi tassi di recidiva dall’anno 1 all’anno 56
TASSO ANNUALIZZATO DI RECIDIVA NEGLI STUDI OPERA I E II
Tasso annualizzato di recidiva aggiustato*
Pazienti trattati con IFN-β-1a 44ug nella DBP
0,4 Pazienti trattati con OCREVUS 600 mg nella fase DBP che hanno
p < 0,001 proseguito con OCREVUS nella OLE
Pazienti passati da IFN-β-1a OCREVUS al basale della OLE
0,3 0,274 p < 0,001
0,203
0,2 p = 0,8
0,14 p = 0,97 p = 0,7
0,125
0,098 0,103
0,1 0,081 0,081 0,072
0,065
0,0
N= 829 827 713 765 623 702 594 665 570 639
Anno 1 Anno 2 Anno 3 Anno 4 Anno 5
(Anno 1 della OLE) (Anno 2 della OLE) (Anno 3 della OLE)
Figura 2, Rif. 6.
*Tasso annualizzato di recidiva aggiustato definito come numero totale di recidive di tutti i pazienti nel gruppo di trattamento diviso
per il numero totale di anni-paziente di esposizione al trattamento. Risultati dell’anno 1 e 2 della DBP relativi alla popolazione
ITT (numero di pazienti disponibili); risultati relativi all’anno 3, 4 e 5 (OLE) relativi alla popolazione ITT della fase di estensione in
aperto (numero di pazienti disponibili).
LO SWITCH DA IFN ß-1A A OCREVUS
ALL’INIZIO DELLA OLE HA DETERMINATO
UNA RIDUZIONE DEL 52% DEL RISCHIO
52% RELATIVO DI ARR A 3 ANNI, CON BENEFICIO
MANTENUTO A 4 E 5 ANNI6
Rate ratio aggiustato
2,069 [IC 95% (1,547-2,766)]
p
La sclerosi multipla è progressiva sin dall’inizio1
SMRR SMSP
Disabilità (EDSS)
Risposta adattiva autoimmune
Infiammazione cronica (microglia)
Demielinizzazione
Transezione/degenerazione
assonale
Volume cerebrale
Soglia
clinica
Soglia
di RM
1 5 10 15 20 anni
SMPP
Figura 1, Rif. 7.
1 5 10 15 20 anni
Dinamiche dei processi clinici e patogenici nella SM. Il grafico in alto mostra l’evoluzione della SM recidivante-remittente
(SMRR) e la sua transizione a SM secondariamente progressiva (SMSP), mentre il grafico in basso mostra l’evoluzione della
SM primariamente progressiva (SMPP). Il processo autoimmune inizia nel sistema immunitario periferico, inducendo episodi
di infiammazione nel sistema nervoso centrale (linea gialla) che, di conseguenza, causa demielinizzazione (linea blu) e poi
degenerazione assonale (linea verde scuro). Sebbene l’infiammazione e la demielinizzazione possano presentare remissione,
la degenerazione assonale si accumula nel tempo, così come l’infiammazione compartimentalizzata (linea arancione).
Se gli infiltrati infiammatori interessano regioni del sistema nervoso centrale significative e superano le soglie di danno,
si manifestano come recidive cliniche. In alternativa, quando la degenerazione assonale cumulativa supera la capacità
di riserva funzionale del sistema nervoso centrale, si origina disabilità neurologica permanente (linea viola) e si osserva
transizione a malattia progressiva. La riduzione del volume cerebrale nel tempo è più grave all’inizio della malattia,
parallelamente alla più intensa attività infiammatoria.
LA SM PUÒ ESSERE CONSIDERATA UNA UNICA PATOLOGIA
PROGRESSIVA NELLA QUALE LE DINAMICHE SPECIFICHE
DI MALATTIA POSSONO DARE LUOGO AD OUTCOME CLINICI
VARI IN DIVERSI GRUPPI DI PAZIENTI.7
EDSS: Expanded Disability Status Scale. SMRR: sclerosis multipla recidivante-remittente. SMSP: sclerosi multipla secondariamente
progressiva. SMPP: sclerosi multipla primariamente progressiva.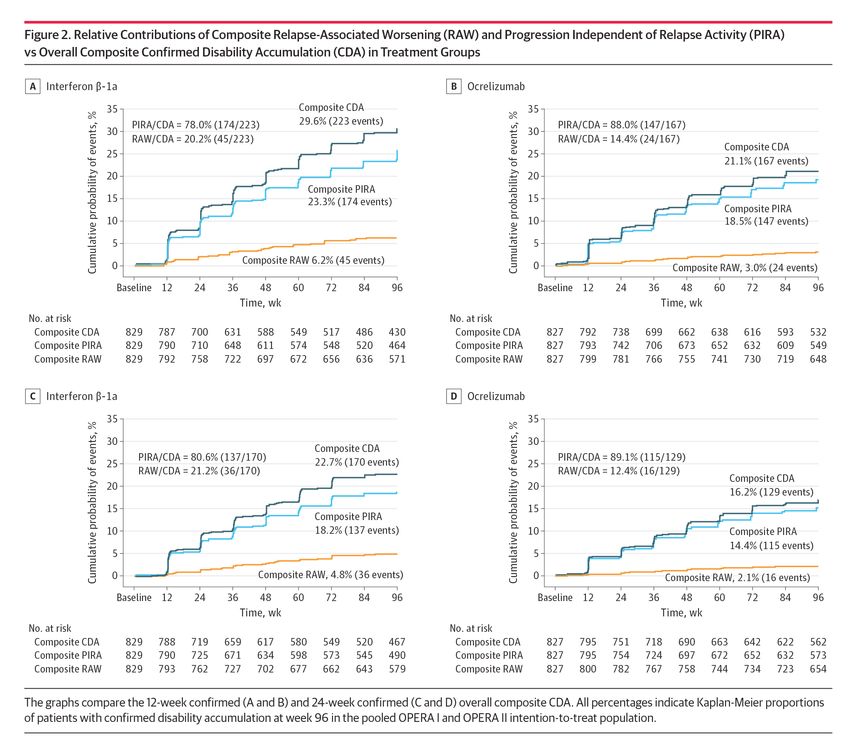
Progressione silente nella sclerosi multipla recidivante in assenza di attività di malattia8 • Il peggioramento a lungo termine è comune nei pazienti affetti da SMRR ed è in gran parte indipendente dalle recidive o dalla formazione di nuove lesioni riscontrabili con la RM cerebrale.8 • Una progressione silente si presenta in molti pazienti con SMRR precoce, che rimangono però classificati come affetti da SM recidivante.8 • È possibile che la perdita di funzionalità nel tempo sia così graduale da passare inosservata al paziente o al medico; in genere, questi pazienti hanno un basso punteggio EDSS e, per la maggior parte, presentano una piena funzionalità.8 • Nello studio EPIC, i pazienti che manifestano progressione silente hanno mostrato un’atrofia cerebrale accelerata a lungo termine e un’atrofia cerebrale aggiustata secondo l’età più elevata al momento dell’arruolamento.8 • Un possibile meccanismo di progressione dei sintomi si basa sulla presenza di lesioni della materia bianca a lenta espansione (SEL) associate a infiammazione cronica al margine anteriore, ma può avere un ruolo anche il danno più diffuso.8 EDSS: Expanded Disability Status Scale. RM: risonanza magnetica. SMRR: sclerosi multipla recidivante-remittente.
Accumulo di disabilità nella SM
Può verificarsi come peggioramento associato a recidiva (RAW)
o come progressione costante, indipendente dalle recidive (PIRA)9
PEGGIORAMENTO ASSOCIATO A RECIDIVA (RAW)
30 gg 30 gg
30 gg 30 gg
10
A 10
basale
rispettoalalbasale
9
9
8
8
EDSS)
7
(punteggioEDSS)
7
relativarispetto
6
6
5
(punteggio
5
4
Disabilitàrelativa
4
3
3
2
Disabilità
2
1 RAW
RAW
1 ≤90 d
≤90 d
Basale Recidiva IID RAW a RAW a
Basale Recidiva IID RAW a RAW a
12 sett. 24 sett.
12 sett. 24 sett.
Tempo (settimane)
Tempo (settimane)
PROGRESSIONE INDIPENDENTE DALL’ATTIVITÀ DI RECIDIVA (PIRA)
30 gg 30 gg 30 gg 30 gg 30 gg
B 7
7
30 gg 30 gg 30 gg 30 gg 30 gg
basale
rispettoalalbasale
6
6
5
EDSS)
5
(punteggioEDSS)
relativarispetto
4
4
(punteggio
3
Disabilitàrelativa
3
2
2 PIRA
PIRA
Disabilità
1
1 ≥30 d
≥30 d
Baseline RelapseRe-baseline IID PIRA a PIRA a
Baseline RelapseRe-baseline IID PIRA a PIRA a
12 sett. 24 sett.
12 sett. 24 sett.
Tempo (settimane)
Tempo (settimane)
Figura 1, Rif. 9.
Rappresentazione schematica delle definizioni di peggioramento associato a recidiva composito (RAW)
e di progressione indipendente dall’attività di recidiva composita (PIRA). Il RAW composito (A) e la PIRA composita (B) sono due
componenti (o driver) non mutualmente esclusivi dell’accumulo complessivo di disabilità misurato con l’accumulo di disabilità
confermata composito (CDA) nelle forme recidivanti o progressive di SM. Il basale dello studio è il punto di riferimento per le
variazioni della disabilità misurate nel tempo; nel contesto degli studi, questo è il momento della randomizzazione al trattamento
in studio, mentre nel contesto clinico questo sarebbe la visita di valutazione della disabilità di riferimento, a partire dalla quale sono
misurate le successivi variazioni nel tempo. Le aree colorate rappresentano gli intervalli intorno alle valutazioni neurologiche che
dovevano restare liberi da recidive per soddisfare il criterio di indipendenza da recidive (all’evento iniziale e ai punti di conferma).
Le valutazioni neurologiche sono state programmate ogni 12 settimane, in accordo con il protocollo dello studio; se si verificava
una recidiva, si effettuava una valutazione neurologica non programmata, nel punto più a sinistra del triangolo della recidiva.
SM: sclerosi multipla. IID: aumento iniziale della disabilità.Accumulo di disabilità indipendente dalle recidive
Nella SM la maggior parte dell’accumulo di disabilità non è associata
a recidiva9
ACCUMULO CONFERMATO DI DISABILITÀ COMPOSITO
A 12 SETTIMANE NEGLI STUDI OPERA I E II
GRUPPO OCREVUS
EVENTI PIRA
88%
(147/167)
Elaborazione grafica di dati da testo, Rif. 9.
GRUPPO IFN β-1a
EVENTI PIRA
78%
(174/223)
Elaborazione grafica di dati da testo, Rif. 9.
IN UNA TIPICA POPOLAZIONE DI PAZIENTI
CON SM RECIDIVANTE, L’80-90% DELL’ACCUMULO
DI DISABILITÀ COMPLESSIVO SI VERIFICA
INDIPENDENTEMENTE DALLE RECIDIVE.9
SM: sclerosi multipla. RWE: peggioramento associato a recidiva. PIRA: progressione indipendente dall’attività di recidiva.
IFN β-1a: interferone beta-1a.Accumulo di disabilità associato o meno alle recidive
OCREVUS è risultato superiore a IFN β-1a nella prevenzione
dell’accumulo confermato di disabilità*, indipendentemente
dalla sua associazione con le recidive9
CONTRIBUTI RELATIVI DEL RAW COMPOSITO E DELLA PIRA COMPOSITA
VS CDA COMPLESSIVA PER GRUPPO DI TRATTAMENTO A 12 SETTIMANE
NEGLI STUDI OPERA I E II
35 35
Probabilità cumulativa dell’evento, %
Probabilità cumulativa dell’evento, %
CDA composito 29,6%
PIRA/CDA = 78,0% (174/223) (223 eventi) PIRA/CDA = 88,0% (147/167)
30 30
RAW/CDA = 20,2% (45/223) RAW/CDA = 14,4% (24/167)
CDA composito
25 25
21,1% (167 eventi)
20 20
PIRA composito
15 15
23,3% (174 eventi) PIRA composito
10 10 18,5% (147 eventi)
5 5
RAW composito 6,2% (45 eventi)
0 0 RAW composito 3,0% (24 eventi)
Basale 12 24 36 48 60 72 84 96 Basale 12 24 36 48 60 72 84 96
Tempo (settimane) Tempo (settimane)
N. a rischio N. a rischio
CDA composito 829 787 700 631 588 549 517 486 430 CDA composito 827 792 738 699 662 638 616 593 532
PIRA composito 829 790 710 648 611 574 548 520 464 PIRA composito 827 793 742 706 673 652 632 609 549
RAW composito 829 792 758 722 697 672 656 636 571 RAW composito 827 799 781 766 755 741 730 719 648
Elaborazione grafica di Figura 2A (sinistra) e 2B (destra), Rif. 9. Originale in allegato.
CON OCREVUS VS IFN β-1a:9
RIDUZIONE RIDUZIONE
DEL RISCHIO DEL RISCHIO
DI PIRA COMPOSITA DI RAW COMPOSITO
A 12 SETTIMANE A 12 SETTIMANE
22% 53%
(HR 0,78; IC 95% 0,63-0,98; p=0,03) (HR 0,47; IC 95% 0,29-0,78; p=0,003)
Elaborazione grafica di dati da testo, Rif. 9.
* Definito come aumento della disabilità rispetto al basale misurato mediante EDSS (aumento di ≥1,0 punto se il valore basale di
EDSS era ≤5,5 punti o aumento di ≥0,5 punti se il valore basale di EDSS era >5,5 punti) o un aumento di almeno il 20%
del T25FW o un aumento di almeno il 20% del 9HPT confermato dopo almeno 12 o almeno 24 settimane.9
IFN β-1a: interferone beta-1a. RWE: peggioramento associato a recidiva. CDA: accumulo di disabilità confermato. PIRA:
progressione indipendente dall’attività di recidiva. HR: hazard ratio.Progressione della disabilità
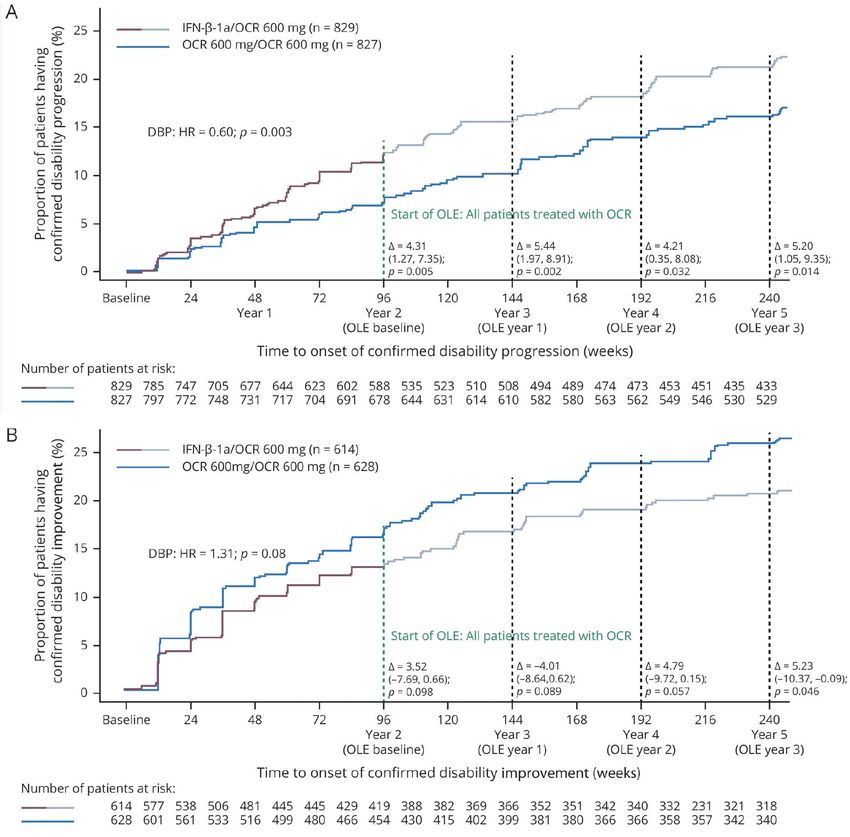
L’analisi dei dati a 5 anni della fase randomizzata in doppio cieco
e dell’estensione in aperto degli studi OPERA suggerisce
che il trattamento precoce e continuativo con OCREVUS determina
benefici che si mantengono nel tempo.6
TEMPO ALL’INSORGENZA DI PROGRESSIONE DELLA DISABILITÀ*
CONFERMATA A 24 SETTIMANE NEGLI STUDI OPERA I E II
25 IFN-β-1a/OCREVUS 600 mg (n=829)
OCREVUS 600 mg/OCREVUS 600 mg (n=827)
Pazienti con progressione della disabilità
20
confermata (%)
15
DBP: HR 0,60; p=0,003
10
5 Inizio della OLE: tutti i pazienti trattati con OCREVUS
Δ = 4,31 Δ = 5,44 Δ = 4,21 Δ = 5,20
(1,27; 7,35) (1,97; 8,91) (0,35; 8,08) (1,05; 9,35)
p = 0,005 p = 0,002 p = 0,032 p = 0,014
0
Basale 24 48 72 96 120 144 168 192 216 240
Anno 1 Anno 2 Anno 3 Anno 4 Anno 5
(Basale della OLE) (Anno 1 della OLE) (Anno 2 della OLE) (Anno 3 della OLE)
Tempo all’insorgenza di progressione della disabilità confermata (settimane)
Numero di pazienti a riscio
829 785 747 705 677 644 623 602 588 535 523 510 508 494 489 474 473 453 451 435 433
827 797 772 748 731 717 704 691 678 644 631 614 610 582 580 563 562 546 546 530 529
Elaborazione grafica di Figura 3A, Rif. 6. Originale in allegato.
Stima di Kaplan-Meier della percentuale di pazienti con eventi di progressione della disabilità confermata
nella fase DBP e nella OLE. Risultati relativi alla popolazione ITT, senza imputazione.
RIDUZIONE DEL RISCHIO DI PROGRESSIONE
DELLA DISABILITÀ CONFERMATA A 24 SETTIMANE
NEI PAZIENTI TRATTATI IN MODO CONTINUATIVO
CON OCREVUS
VS SWITCH A IFN β-1a NELLA FASE IN DOPPIO CIECO6
-40%
(HR 0,60; IC 95% 0,43-0,84; p=0,003)
Elaborazione grafica di dati da testo, Rif. 6.
* Definita come un aumento rispetto al basale del punteggio EDSS di almeno 1 punto (o di 0,5 punti se il valore basale
di EDSS era >5,5) mantenuto per almeno 12 settimane.10
DBP: fase controllata in doppio cieco. OLE: estensione in aperto. IFN β-1a: interferone beta-1a. ITT: intention-to-treat.
HR: hazard ratio. EDSS: Expanded Disability Status Scale.Miglioramento della disabilità
OCREVUS è associato a più elevate percentuali di miglioramento
della disabilità* confermata a 12 settimane rispetto a IFN β-1a10
PAZIENTI CON MIGLIORAMENTO DELLA DISABILITÀ CONFERMATO
DOPO ALMENO 12 SETTIMANE NEGLI STUDI OPERA I E II
25
Pazienti con miglioramento della disabilità confermato
20
20.7
di 12 settimane alla settimana 96 (%)
AUMENTO
DEL
15
33%
vs IFN-β-1a
15.6
10
5
0
IFN β-1a OCREVUS
44 μg 600 mg
(n=614) (n=628)
Figura S5, Rif. 11.
Analisi raggruppata della proporzione di pazienti che hanno ottenuto il miglioramento della disabilità confermato dopo 12
settimane. CDI indica il miglioramento della disabilità confermato e IFN l’interferone.
*Definito come riduzione del punteggio EDSS rispetto al basale di almeno 1 punto (o 0,5 punti se il punteggio EDSS al basale era
>5,5) mantenuto per almeno 12 settimane nei pazienti con punteggio EDSS al basale di almeno 2,0.10
IFN β-1a: interferone beta-1a.OCREVUS: efficacia dimostrata
sulla progressione della disabilità
TWO IDENTICAL 2-YEAR TRIALS
PREVENZIONE DEL CDA
INDIPENDENTE DALLE RECIDIVE9§
BENEFICI A LUNGO TERMINE
DEL TRATTAMENTO PRECOCE6+
SUPERIORE MIGLIORAMENTO DELLA DISABILITÀ10‡
Bibliografia:
1. Andersson PB et al. Multiple sclerosis that is progressive from the time of onset: clinical characteristics and progression of disability.
Arch Neurol. 1999 Sep;56(9):1138-42. 2. Merkel B et al. Timing of high-efficacy therapy in relapsing-remitting multiple sclerosis:
a systematic review. Autoimmun Rev. 2017 Jun;16(6)658-665. 3. Coetzee T, Thompson AJ. Unified understanding of MS course is
required for drug development. Nat Rev Neurol. 2018 Apr;14(4):191-192. 4. Reich DS et al. Multiple Sclerosis. N Engl J Med. 2018
Jan 11;378(2):169-180. 5. OCREVUS®, Riassunto delle caratteristiche del prodotto. 6. Hauser SL et al. Five years of ocrelizumab
in relapsing multiple sclerosis. OPERA studies open-label extension. Neurology 2020;95(13):e1854-e1867. 7. Kotelnikova E et al.
Dynamics and heterogeneity of brain damage in multiple sclerosis. PLoS Comput Biol. 2017;26(10):e1005757. doi:10.1371/journal.
pcbi.1005757. 8. Cree B et al. Silent Progression in Disease Activity -Free Relapsing Multiple Sclerosis. Ann Neurol 2019;85:653 -666
9. Kappos L et al. Contribution of Relapse-Independent Progression vs Relapse-Associated Worsening to Overall Confirmed
Disability Accumulation in Typical Relapsing Multiple Sclerosis in a Pooled Analysis of 2 Randomized Clinical Trials 2020. JAMA
Neurol 2020;77(9):1-9. doi: 10.1001/jamaneurol.2020.1568. 10. Hauser SL et al. Ocrelizumab versus Interferon Beta-1a in Relapsing
Multiple Sclerosis. N Engl J Med 2017;376:221-34. 11. Hauser SL et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple
Sclerosis. N Engl J Med 2017;376:221-34. Supplementary Appendix.
Ocrevus - 300 mg concentrato per soluzione per infusione
1 flaconcino da 10 ml AIC n° 045889019/E
Classificazione ai fini di rimborsabilità/regime di fornitura: H/OSP
Prezzo al pubblico (IVA inclusa): € 9.309,29*
* Prezzi al netto delle riduzioni temporanee (Suppl. nr. 161 alla G.U. nr. 156 del 7 luglio 2006 e G.U.
nr. 227 del 29 settembre 2006) e aggiornati secondo quanto disposto della normativa relativa
al pay-back (determinazione AIFA 28 febbraio 2007) e successive proroghe.
§
OCREVUS è risultato superiore a IFN β-1a nella prevenzione dell’accumulo confermato di disabilità, indipendentemente
dalla sua associazione con le recidive.9
†
L’analisi dei dati a 5 anni della fase randomizzata in doppio cieco e dell’estensione in aperto degli studi OPERA suggerisce
che il trattamento precoce e continuativo con OCREVUS determina benefici che si mantengono nel tempo.6
‡
OCREVUS è associato a più elevate percentuali di miglioramento della disabilità confermata
a 12 settimane rispetto a IFN β-1a.10
IFN β-1a: interferone beta-1a. CDA: accumulo di disabilità confermato.
RCP Incluso. Cod. M-IT-00000350
Depositato presso AIFA in data 15/11/2020Immagini originali Fig. 3 da Rif. 6
Immagini originali Fig. 2 da Rif. 9
▼
RIASSUNTO DELLE CARATTERISTICHE DEL PRODOTTO Se si osservano segni di una IRR potenzialmente letale o invalidante durante un’infu-
▼ Medicinale sottoposto a monitoraggio addizionale. Ciò permetterà la rapida iden- sione, come ipersensibilità acuta o sindrome da distress respiratorio acuto, l’infusione
tificazione di nuove informazioni sulla sicurezza. Agli operatori sanitari è richiesto di deve essere interrotta immediatamente e il paziente deve ricevere un trattamento
segnalare qualsiasi reazione avversa sospetta. Vedere paragrafo 4.8 per informazioni appropriato. In questi pazienti Ocrevus deve essere sospeso in modo permanente
sulle modalità di segnalazione delle reazioni avverse. (vedere paragrafo 4.3).
1. DENOMINAZIONE DEL MEDICINALE IRR severa
Ocrevus 300 mg concentrato per soluzione per infusione Se un paziente manifesta una IRR severa (per es. dispnea) o un insieme di sintomi
2. COMPOSIZIONE QUALITATIVA E QUANTITATIVA quali vampate di calore, febbre e mal di gola, l’infusione deve essere interrotta imme-
Ogni flaconcino contiene 300 mg di ocrelizumab in 10 mL a una concentrazione di diatamente e il paziente deve ricevere un trattamento sintomatico. L’infusione deve
30 mg/mL. La concentrazione finale del farmaco dopo diluizione è di circa 1,2 mg/mL. essere ripresa soltanto dopo che tutti i sintomi si sono risolti. Alla ripresa la velocità di
Ocrelizumab è un anticorpo monoclonale ricombinante umanizzato anti-CD20 prodot- infusione iniziale deve essere la metà della velocità di infusione utilizzata al momento
to da cellule di ovaio di hamster cinese mediante la tecnologia del DNA ricombinante. dell’insorgenza della reazione. Per le successive nuove infusioni non è necessario
Per l’elenco completo degli eccipienti, vedere paragrafo 6.1. attuare alcuna modifica dell’infusione, a meno che il paziente manifesti una IRR.
3. FORMA FARMACEUTICA IRR da lieve a moderata
Concentrato per soluzione per infusione. Se un paziente manifesta una IRR da lieve a moderata (per es. cefalea), la velocità
Soluzione di aspetto da limpido a leggermente opalescente, da incolore a marrone di infusione deve essere ridotta e portata a metà della velocità utilizzata al momento
chiaro. dell’insorgenza dell’evento. Questa velocità ridotta deve essere mantenuta per almeno
4. INFORMAZIONI CLINICHE 30 minuti. Se tollerata, la velocità di infusione potrà essere aumentata in base alla ve-
4.1 Indicazioni terapeutiche locità di infusione iniziale del paziente. Per le successive nuove infusioni non è neces-
Ocrevus è indicato per il trattamento di pazienti adulti affetti da forme recidivanti di sario attuare alcuna modifica dell’infusione, a meno che il paziente manifesti una IRR.
sclerosi multipla (SMR) con malattia attiva definita in base alle caratteristiche cliniche Modifiche della posologia durante il trattamento
o radiologiche (vedere paragrafo 5.1). I suddetti esempi di interruzione e rallentamento della somministrazione della dose
Ocrevus è indicato per il trattamento di pazienti adulti affetti da sclerosi multipla pri- (per IRR lievi/moderate e severe) determineranno una modifica della velocità di infu-
mariamente progressiva (SMPP) in fase precoce in termini di durata della malattia e sione e un aumento della durata complessiva dell’infusione, ma non della dose totale.
livello di disabilità, con caratteristiche radiologiche tipiche di attività infiammatoria Non sono raccomandate riduzioni della dose di Ocrevus.
(vedere paragrafo 5.1). Dosi ritardate o dimenticate
4.2 Posologia e modo di somministrazione Se si dimentica un’infusione di Ocrevus, questa dovrà essere somministrata il prima
Il trattamento con Ocrevus deve essere iniziato e supervisionato da un medico spe- possibile; non si deve attendere la successiva dose programmata. Tra una dose e
cializzato, esperto nella diagnosi e nel trattamento di condizioni neurologiche e che l’altra di Ocrevus si deve mantenere l’intervallo di trattamento di 6 mesi (minimo 5
abbia accesso a idonee misure di supporto medico per gestire reazioni severe, come mesi; vedere Tabella 1).
le reazioni gravi correlate all’infusione (Infusion-Related Reaction, IRR). Popolazioni speciali
Premedicazione per reazioni correlate all’infusione Adulti di età superiore a 55 anni e popolazione anziana
Prima di ogni infusione di Ocrevus si devono somministrare le seguenti due premedi- In base ai dati limitati disponibili (vedere paragrafo 5.1 e paragrafo 5.2), non è neces-
cazioni allo scopo di ridurre la frequenza e la severità delle IRR (per ulteriori indicazioni saria alcuna correzione della posologia nei pazienti di età superiore a 55 anni. Dopo
su come ridurre le IRR, vedere “Reazioni correlate all’infusione” nel paragrafo 4.4). aver compiuto 55 anni di età, i pazienti arruolati negli studi clinici in corso continuano
• 100 mg di metilprednisolone (o un equivalente) per via endovenosa circa 30 mi- a essere trattati con ocrelizumab 600 mg ogni sei mesi.
nuti prima di ciascuna infusione di Ocrevus; Alterazione della funzionalità renale
• antistaminico circa 30-60 minuti prima di ciascuna infusione di Ocrevus. La sicurezza e l’efficacia di Ocrevus in pazienti con funzionalità renale alterata non
Si può inoltre valutare l’opportunità di somministrare una premedicazione con un an- sono state oggetto di studi specifici. Pazienti con lieve compromissione della funzio-
tipiretico (per es. paracetamolo) circa 30-60 minuti prima di ciascuna infusione di nalità renale sono stati inclusi negli studi clinici. Non vi sono esperienze in pazienti con
Ocrevus. compromissione della funzionalità renale moderata e severa. Ocrevus è un anticorpo
Posologia: monoclonale ed è eliminato mediante catabolismo (ossia degradazione in peptidi e
Dose iniziale aminoacidi) e non si prevede la necessità di modifiche posologiche nei pazienti con
La dose iniziale di 600 mg è somministrata mediante due diverse infusioni endove- compromissione della funzionalità renale (vedere paragrafo 5.2).
nose: una prima infusione da 300 mg, seguita da una seconda infusione da 300 mg Alterazione della funzionalità epatica
2 settimane più tardi (Tabella 1). La sicurezza e l’efficacia di Ocrevus in pazienti con compromissione epatica non sono
Dosi successive state oggetto di studi specifici. Pazienti con una compromissione epatica lieve sono
In seguito le dosi successive di Ocrevus vengono somministrate mediante singola stati inclusi negli studi clinici. Non vi sono esperienze in pazienti con compromissione
infusione endovenosa da 600 mg ogni 6 mesi (Tabella 1). La prima dose successiva epatica moderata e severa. Ocrevus è un anticorpo monoclonale ed è smaltito me-
da 600 mg deve essere somministrata sei mesi dopo la prima infusione della dose diante catabolismo (piuttosto che mediante metabolismo epatico) e non si prevede la
iniziale. Se durante le precedenti infusioni di Ocrevus i pazienti non hanno manifesta- necessità di modifiche posologiche nei pazienti con compromissione epatica (vedere
to una grave reazione correlata all’infusione (Infusion-Related Reaction, IRR), le dosi paragrafo 5.2).
successive possono essere somministrate con un’infusione in tempi ridotti (di 2 ore Popolazione pediatrica
di durata) (Tabella 1, Opzione 2). Si deve mantenere un intervallo minimo di 5 mesi Nei bambini e negli adolescenti da 0 a 18 anni di età la sicurezza e l’efficacia di Ocre-
tra le dosi di Ocrevus. vus non sono state ancora stabilite. Non ci sono dati disponibili.
Modifiche dell’infusione in caso di IRR Metodo di somministrazione
In caso di IRR durante un’infusione, procedere con i seguenti aggiustamenti. Ulteriori Dopo diluizione, Ocrevus è somministrato mediante infusione endovenosa attraverso
informazioni sulle IRR sono riportate nel paragrafo 4.4. una linea dedicata. Ocrevus non deve essere somministrato come push o bolo endo-
IRR potenzialmente letale venoso.
Tabella 1. Posologia di Ocrevus
Quantità di
Istruzioni
Ocrevus
per l’infusione
da somministrare
Dose iniziale Infusione 1 300 mg in 250 mL • Iniziare l’infusione a una velocità di 30 mL/ora per 30 minuti
(600 mg) Infusione 2 300 mg in 250 mL • La velocità può essere aumentata con incrementi da 30 mL/ora ogni 30 mi-
Suddivisa in 2 infusioni (2 settimane più tardi) nuti fino a un massimo di 180 mL/ora
• Ogni infusione deve essere somministrata nell’arco di circa 2,5 ore
Dosi successive Opzione 1 600 mg in 500 mL • Iniziare l’infusione a una velocità di 40 mL/ora per 30 minuti
(600 mg) Infusione di durata • La velocità può essere aumentata con incrementi da 40 mL/ora ogni 30 mi-
Infusione singola pari a circa 3,5 ore nuti fino a un massimo di 200 ml/ora
una volta ogni 6 mesi • Ogni infusione deve essere somministrata nell’arco di circa 3,5 ore
Oppure
Opzione 2 600 mg in 500 mL • Iniziare l’infusione a una velocità di 100 mL/ora per i primi 15 minuti
Infusione di durata • Aumentare la velocità di infusione a 200 mL/ora per i successivi 15 minuti
pari a circa 2 ore • Aumentare la velocità di infusione a 250 mL/ora per i successivi 30 minuti
• Aumentare la velocità di infusione a 300 mL/ora per i restanti 60 minuti
• Ogni infusione deve essere somministrata nell’arco di circa 2 oreLe soluzioni di Ocrevus per infusione endovenosa si preparano diluendo il medicinale popolazione di pazienti, politerapia con immunosoppressori). I medici devono prestare
in una sacca per infusione contenente sodio cloruro allo 0,9% fino a raggiungere una attenzione ai primi segni e sintomi di PML, che possono includere qualsiasi nuova
concentrazione finale di circa 1,2 mg/mL. Per istruzioni sulla diluizione del medicinale insorgenza o peggioramento di segni e sintomi neurologici, poiché questi possono es-
prima della somministrazione, vedere paragrafo 6.6. I pazienti devono essere moni- sere simili alla SM. In caso di sospetta PML, si deve sospendere la somministrazione di
torati durante l’infusione e per almeno un’ora dopo il completamento dell’infusione Ocrevus. Si deve quindi valutare l’opportunità di eseguire accertamenti, compresa una
(vedere paragrafo 4.4). risonanza magnetica (RM) preferibilmente con contrasto (da confrontare con la RM
4.3 Controindicazioni pre-trattamento), l’analisi di conferma del liquido cerebrospinale (LCS) per ricercare
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al para- l’acido desossiribonucleico (DNA) del virus di John Cunningham (JC) e la ripetizione
grafo 6.1. degli esami neurologici. Se la PML è confermata, il trattamento deve essere interrotto
• Infezione attiva in corso (vedere paragrafo 4.4) in via definitiva.
• Pazienti in stato severamente immunocompromesso (vedere paragrafo 4.4) Riattivazione dell’epatite B
• Neoplasie maligne attive note (vedere paragrafo 4.4) La riattivazione del virus dell’epatite B (HBV), che in alcuni casi determina epatite
4.4 Avvertenze speciali e precauzioni di impiego fulminante, insufficienza epatica e morte, è stata riferita in pazienti trattati con anti-
Tracciabilità corpi anti-CD20. Lo screening per l’HBV deve essere eseguito in tutti i pazienti prima
Per migliorare la tracciabilità dei medicinali biologici il nome e numero di lotto del dell’inizio del trattamento con Ocrevus secondo le linee guida locali. I pazienti con HBV
prodotto somministrato devono essere chiaramente registrati. attivo (ossia un’infezione confermata da risultati positivi dei test HBsAg e anti-HB) non
Reazioni correlate all’infusione (IRR) devono essere trattati con Ocrevus. I pazienti con sierologia positiva (ossia con risultati
Ocrevus si associa a IRR, che possono essere correlate al rilascio di citochine e/o a di negativi per HBsAg e positivi per l’anticorpo core dell’HB (HBcAb +) e i portatori di HBV
altri mediatori chimici. I sintomi di IRR possono presentarsi durante qualsiasi infusio- (positivi per l’antigene di superficie, HBsAg+) devono consultare un esperto in malattie
ne, ma sono stati riferiti con maggiore frequenza durante la prima infusione. Le IRR epatiche prima di iniziare il trattamento e devono essere monitorati e gestiti ai sensi
possono manifestarsi entro 24 ore dall’infusione. Tali reazioni possono presentarsi in degli standard medici locali per prevenire una riattivazione dell’epatite B.
forma di prurito, rash cutaneo, orticaria, eritema, irritazione della gola, dolore orofarin- Neoplasie maligne
geo, dispnea, edema faringeo o laringeo, vampate, ipotensione, piressia, affaticamen- Negli studi clinici è stato riferito un numero aumentato di neoplasie maligne (tra cui
to, cefalea, capogiri, nausea e tachicardia (vedere paragrafo 4.8). carcinomi mammari) nei pazienti trattati con ocrelizumab rispetto ai gruppi di con-
Prima dell’infusione: trollo. L’incidenza rientrava tuttavia nel range di riferimento atteso per la popolazione
• Gestione delle reazioni severe: si deve disporre di mezzi adeguati per gestire le con SM. Nei pazienti con fattori di rischio noti per lo sviluppo di neoplasie maligne e
reazioni severe, come IRR gravi, reazioni di ipersensibilità e/o reazioni anafilat- in quelli sottoposti a monitoraggio attivo per recidiva di neoplasia maligna si deve
tiche. prendere in considerazione il rapporto beneficio/rischio individuale. I pazienti con ne-
• Ipotensione: può verificarsi come sintomo di una IRR durante le infusioni di Ocre- oplasia maligna attiva nota non devono essere trattati con Ocrevus (vedere paragrafo
vus. Pertanto, nelle 12 ore precedenti ciascuna infusione di Ocrevus e durante 4.3). I pazienti devono seguire lo screening standard per il carcinoma mammario in
l’infusione stessa si deve valutare l’opportunità di sospendere i trattamenti an- funzione delle linee guida locali. Per le popolazioni non studiate, vedere paragrafo 4.2.
tipertensivi. I pazienti con anamnesi di insufficienza cardiaca congestizia (New Nel periodo controllato degli studi clinici, l’incidenza di carcinomi cutanei diversi dal
York Heart Association III e IV) non sono stati studiati. melanoma è risultata bassa e non è stata osservata alcuna sproporzione tra i gruppi di
• Premedicazione: i pazienti devono ricevere una premedicazione per ridurre la trattamento. Tra gli anni 3 e 4 del trattamento è stato registrato un aumento dell’inci-
frequenza e la gravità delle IRR (vedere paragrafo 4.2). denza imputabile al carcinoma basocellulare, che non è stato osservato negli anni suc-
Durante l’infusione: cessivi. L’incidenza rientra nel range di riferimento atteso per la popolazione con SM.
• Nei pazienti che manifestano sintomi polmonari severi, come broncospasmo o Trattamento dei pazienti severamente immunocompromessi
esacerbazione dell’asma, devono essere adottate le seguenti misure: I pazienti in stato severamente immunocompromesso non devono essere trattati fino
- si deve interrompere l’infusione immediatamente e in via definitiva a quando la condizione non si risolva (vedere paragrafo 4.3).
- deve essere somministrato un trattamento sintomatico In altre patologie autoimmuni l’uso di Ocrevus in concomitanza con medicinali im-
- si deve monitorare il paziente fino alla risoluzione dei sintomi polmonari poi- munosoppressori (per es. terapia cronica con corticosteroidi, farmaci antireumatici
ché un iniziale miglioramento dei sintomi clinici può essere seguito da un modificanti la malattia [Disease Modifying Antirheumatic Drug, DMARD] biologici e
peggioramento. non biologici, micofenolato mofetile, ciclofosfamide, azatioprina) ha determinato un
• Potrebbe essere difficile distinguere i sintomi di ipersensibilità da quelli di una incremento di infezioni gravi, incluse infezioni opportunistiche. Le infezioni rilevate
IRR. In caso di sospetta reazione di ipersensibilità durante l’infusione, l’infusione hanno incluso, a titolo esemplificativo ma non esaustivo, polmonite atipica e polmoni-
deve essere interrotta immediatamente e in via definitiva (vedere di seguito “Re- te da pneumocystis jirovecii, polmonite da varicella, tubercolosi, istoplasmosi. In rari
azioni di ipersensibilità”). casi alcune di queste infezioni hanno avuto un esito fatale. Un’analisi esplorativa ha
Dopo l’infusione: identificato i seguenti fattori associati al rischio di infezioni gravi: dosi di Ocrevus più
• I pazienti trattati con Ocrevus devono essere posti sotto osservazione per almeno elevate di quanto raccomandato per la SM, altre comorbilità e uso cronico di immu-
un’ora dopo il completamento dell’infusione per rilevare eventuali sintomi di IRR. nosoppressori/corticosteroidi. L’uso di altri immunosoppressori in concomitanza con
• I medici devono avvisare i pazienti della possibilità che una IRR si verifichi nelle Ocrevus non è raccomandato, eccetto per i corticosteroidi utilizzati per il trattamento
24 ore successive all’infusione. sintomatico di recidive. Si dispone di conoscenze limitate in merito alla possibilità che
Vedere paragrafo 4.2 per una guida sulla posologia nei pazienti che manifestano sin- l’uso concomitante di steroidi per il trattamento sintomatico delle recidive sia asso-
tomi di una IRR. ciato a un aumento del rischio di infezioni nella pratica clinica. Negli studi registrativi
Reazioni di ipersensibilità condotti con ocrelizumab nella SM, la somministrazione di corticosteroidi per il trat-
Può manifestarsi anche una reazione di ipersensibilità (reazione allergica acuta a un tamento delle recidive non si è associata a un aumento del rischio di infezioni gravi.
medicinale). Le reazioni di ipersensibilità acute di tipo I (IgE-mediate) potrebbero es- Quando si inizia il trattamento con Ocrevus dopo una terapia immunosoppressiva o
sere indistinguibili dal punto di vista clinico dai sintomi delle IRR. quando si inizia una terapia immunosoppressiva dopo Ocrevus, si deve considerare
Una reazione di ipersensibilità può presentarsi durante qualsiasi infusione, ma in ge- la possibilità di sovrapposizione degli effetti farmacodinamici (vedere paragrafo 5.1
nere non si presenta durante la prima infusione. Nelle infusioni successive la mani- Effetti farmacodinamici). Occorre osservare la dovuta cautela nel prescrivere Ocrevus
festazione di sintomi più severi di quelli manifestati in precedenza o di nuovi sintomi e prendere in considerazione la farmacodinamica delle altre terapie per la SM modi-
severi deve indurre a valutare la possibilità di una reazione di ipersensibilità. I pazienti ficanti la malattia.
con nota ipersensibilità a ocrelizumab IgE-mediata non devono essere trattati (vedere Vaccinazioni
paragrafo 4.3). La sicurezza dell’immunizzazione con vaccini vivi o vivi attenuati dopo la terapia con
Infezione Ocrevus non è stata studiata e la vaccinazione con vaccini vivi o vivi attenuati non è
La somministrazione di Ocrevus deve essere posticipata nei pazienti con un’infezione raccomandata durante il trattamento e fino a ricostituzione delle cellule B (negli studi
attiva fino alla risoluzione della stessa. clinici, il tempo mediano alla ricostituzione delle cellule B è stato di 72 settimane).
Prima della somministrazione si raccomanda di verificare lo stato immunitario del pa- Vedere paragrafo 5.1. In uno studio randomizzato in aperto, i pazienti affetti da SMR
ziente, in quanto i pazienti severamente immunocompromessi (per es. con linfopenia, sono stati in grado di produrre risposte umorali, anche se ridotte, al vaccino con tos-
neutropenia, ipogammaglobulinemia) non devono essere trattati (vedere paragrafi 4.3 soide tetanico, al vaccino antipneumococcico polisaccaridico 23-valente con o senza
e 4.8). La percentuale complessiva di pazienti che hanno manifestato un’infezione richiamo, al vaccino con il neoantigene emocianina di Megathura crenulata e al vacci-
grave è risultata simile a quella osservata con i medicinali di confronto (vedere para- no antinfluenzale stagionale. Vedere paragrafi 4.5 e 5.1. Si raccomanda di vaccinare i
grafo 4.8). La frequenza delle infezioni di grado 4 (potenzialmente letali) e 5 (fatali) si è pazienti trattati con Ocrevus con vaccini antinfluenzali stagionali inattivati. Nel consi-
rivelata bassa in tutti i gruppi di trattamento, ma nella SMPP è risultata superiore con derare il trattamento con Ocrevus i medici devono valutare lo stato di immunizzazione
Ocrevus rispetto al placebo per infezioni potenzialmente letali (1,6% versus 0,4%) e del paziente. I pazienti che necessitano di una vaccinazione devono completare la
infezioni fatali (0,6% versus 0%). Tutte le infezioni potenzialmente letali si sono risolte propria immunizzazione almeno 6 settimane prima di iniziare la terapia con Ocrevus.
senza interrompere la somministrazione di ocrelizumab. I pazienti affetti da SMPP con Per maggiori informazioni sulle vaccinazioni, vedere paragrafi 4.5 e 5.1.
difficoltà di deglutizione sono maggiormente esposti al rischio di polmonite ab inge- Esposizione a ocrelizumab in utero e vaccinazioni nei neonati e nei lattanti con vaccini
stis. In questi pazienti, il trattamento con Ocrevus può incrementare ulteriormente il vivi o vivi attenuati
rischio di polmonite severa. I medici devono intervenire tempestivamente nei pazienti A causa della potenziale deplezione delle cellule B nei lattanti di madri che sono state
che manifestano polmonite. esposte a Ocrevus durante la gravidanza, si raccomanda di posticipare la vaccina-
Leucoencefalopatia multifocale progressiva (PML) zione con vaccini vivi o vivi attenuati fino a recupero dei livelli di cellule B; pertanto,
Non si può escludere un rischio di PML poiché l’infezione da virus di John Cunningham prima della vaccinazione, si raccomanda di misurare nei neonati e nei lattanti i livelli
(JC) con conseguente sviluppo di PML è stata osservata in pazienti trattati con anticor- di cellule B CD19-positive. Si raccomanda che tutte le vaccinazioni diverse da quelle
pi anti-CD20 e con altre terapie per la SM, e si è associata a fattori di rischio (per es. con vaccini vivi o vivi attenuati seguano il programma di immunizzazione locale. Perverificare che i soggetti abbiano prodotto una risposta immunitaria protettiva è ne- vivi attenuati. Nei neonati e nei lattanti esposti a ocrelizumab non sono stati raccolti
cessario prendere in considerazione la misurazione dei titoli delle risposte anticorpali dati sulla conta delle cellule B e non è nota la potenziale durata della deplezione delle
indotte dal vaccino, in quanto l’efficacia della vaccinazione potrebbe essere ridotta. cellule B in queste popolazioni (vedere paragrafo 4.4).
La sicurezza e la tempistica delle vaccinazioni devono essere discusse con il pediatra In neonati nati da madri esposte ad altri anticorpi anti-CD20 durante la gravidanza
(vedere paragrafo 4.6). sono state riferite deplezioni transitorie delle cellule B periferiche e linfocitopenia.
Sodio Gli studi sugli animali (tossicità embrio-fetale) non indicano effetti teratogeni. È stata
Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per dose, ossia è so- rilevata una deplezione delle cellule B in utero. Gli studi sullo sviluppo pre- e post-na-
stanzialmente privo di sodio. tale hanno mostrato tossicità riproduttiva (vedere paragrafo 5.3).
4.5 Interazioni con altri medicinali e altre forme di interazione Ocrevus deve essere evitato durante la gravidanza a meno che il potenziale beneficio
Non sono stati effettuati studi formali di interazione farmacologica, poiché non si per la madre superi il potenziale rischio per il feto.
attendono interazioni farmacologiche attraverso gli enzimi del citocromo P450 o altri Allattamento
enzimi metabolizzanti o trasportatori. Non è noto se i metaboliti di ocrelizumab siano escreti nel latte materno. I dati
Vaccinazioni farmacodinamici/tossicologici disponibili sugli animali hanno mostrato l’escre-
La sicurezza dell’immunizzazione con vaccini vivi o vivi attenuati dopo la terapia con zione di ocrelizumab nel latte (per maggiori dettagli, vedere paragrafo 5.3). Non
Ocrevus non è stata studiata. Sono disponibili dati sugli effetti del vaccino con tos- può essere esclusa la sussistenza di un rischio per i neonati e i lattanti. Le donne
soide tetanico, del vaccino antipneumococcico polisaccaridico 23-valente (23-PPV), devono essere invitate a interrompere l’allattamento al seno durante la terapia
del vaccino con il neoantigene emocianina di Megathura crenulata e del vaccino con Ocrevus.
antinfluenzale stagionale in pazienti trattati con Ocrevus. Vedere paragrafi 4.4 e 5.1. Fertilità
Dopo 2 anni di trattamento con Ocrevus, la proporzione di pazienti con titoli positivi I dati preclinici non rivelano speciali pericoli per l’uomo sulla base degli studi di
degli anticorpi contro S. pneumoniae, parotite, rosolia e varicella è stata generalmen- fertilità maschile e femminile condotti in scimmie cynomolgus.
te simile alle proporzioni basali. 4.7 Effetti sulla capacità di guidare veicoli e sull’uso di macchinari
Immunosoppressori Ocrevus non altera o altera in modo trascurabile la capacità di guidare veicoli e di
È sconsigliato usare altri immunosoppressori in concomitanza con Ocrevus, fatta usare macchinari.
eccezione per i corticosteroidi per il trattamento sintomatico delle recidive. 4.8 Effetti indesiderati
Per informazioni sull’uso degli immunosoppressori prima, durante o dopo il tratta- Riassunto del profilo di sicurezza
mento con Ocrevus, vedere paragrafo 4.4 “Trattamento dei pazienti severamente Le reazioni avverse al farmaco (ADR) più importanti e segnalate con maggiore fre-
immunocompromessi”. quenza sono state IRR e infezioni. Per ulteriori informazioni, vedere paragrafi 4.4 e
4.6 Fertilità, gravidanza e allattamento 4.8 (sottoparagrafo “Descrizione di reazioni avverse selezionate”).
Donne potenzialmente fertili Tabella delle reazioni avverse
Le donne potenzialmente fertili devono utilizzare misure contraccettive durante la Il profilo di sicurezza complessivo di Ocrevus nella sclerosi multipla si basa sui dati in
terapia con Ocrevus e per 12 mesi dopo l’ultima infusione di Ocrevus (vedere di pazienti arruolati negli studi clinici registrativi nella SM (SMR e SMPP).
seguiti paragrafi 5.1 e 5.2). La Tabella 2 riassume le ADR che sono state riferite in associazione all’uso di Ocrevus
Gravidanza in 1311 pazienti (3054 anni-paziente) durante i periodi di trattamento controllato
Ocrevus è un anticorpo monoclonale umanizzato di un sottotipo di immunoglobulina degli studi clinici nella SM.
G1 ed è noto che le immunoglobuline attraversano la barriera placentare. Le frequenze sono definite come: molto comune (≥ 1/10), comune (≥ 1/100 e
I dati relativi all’uso di Ocrevus in donne in gravidanza sono in numero limitato. Nei < 1/10), non comune (≥ 1/1.000 e < 1/100), raro (≥ 1/10.000 e < 1/1.000) e molto
neonati e nei lattanti nati da madri esposte ad Ocrevus durante la gravidanza va raro (≤ 1/10.000). All’intero di ciascun raggruppamento per sistemi e organi, le rea-
presa in considerazione la possibilità di posticipare la vaccinazione con vaccini vivi o zioni avverse sono presentate in ordine decrescente di frequenza.
Tabella 2. ADR riportate con Ocrevus (nella SMR o SMPP)
MedDRA Molto comune Comune
Classificazione per sistemi e organi
Infezioni e infestazioni Infezione delle vie aeree superiori, Sinusite, bronchite, herpes orale, gastroenterite, infezione delle
rinofaringite, influenza vie aeree, infezione virale, herpes zoster, congiuntivite, cellulite
Patologie respiratorie, toraciche e mediastiniche Tosse, catarro
Esami diagnostici Riduzione dei livelli ematici Riduzione dei livelli ematici di immunoglobulina G
di immunoglobulina M
Patologie del sistema emolinfopoietico Neutropenia
Traumatismo, avvelenamento e complicazioni da procedura Reazioni correlate all’infusione 1
1
I sintomi segnalati come IRR nelle 24 ore successive all’infusione sono descritti di seguito nel paragrafo “Reazioni correlate all’infusione”.
Descrizione di reazioni avverse selezionate ore) in pazienti con sclerosi multipla recidivante-remittente, l’incidenza, l’intensità e la
Reazioni correlate all’infusione tipologia dei sintomi delle IRR sono risultati in linea con quelli delle infusioni sommini-
Nei diversi studi sulla SMR e sulla SMPP, i sintomi associati a IRR hanno incluso, strate nell’arco di 3,5 ore (vedere paragrafo 5.1 Proprietà farmacodinamiche). Il numero
a titolo esemplificativo ma non esaustivo: prurito, rash cutaneo, orticaria, eritema, complessivo di interventi necessari è risultato basso in entrambi i gruppi di infusione,
vampate, ipotensione, piressia, affaticamento, cefalea, capogiri, irritazione della gola, tuttavia, sono risultati necessari più interventi (rallentamento o interruzioni temporanee)
dolore orofaringeo, dispnea, edema faringeo o laringeo, nausea, tachicardia. Negli per gestire le IRR nel gruppo di infusione in tempi ridotti (2 ore) rispetto al gruppo di
studi controllati non sono state registrate IRR con esito fatale. infusione somministrata in 3,5 ore (8,7% contro il 4,8%, rispettivamente).
Negli studi clinici con controllo attivo (SMR), le IRR hanno rappresentato l’evento Infezione
avverso più comune nei pazienti trattati con Ocrevus, con un’incidenza complessiva Negli studi con controllo attivo sulla SMR si sono manifestate infezioni nel 58,5% dei
del 34,3% rispetto a un’incidenza del 9,9% nel gruppo trattato con interferone be- pazienti trattati con Ocrevus e nel 52,5% di quelli trattati con interferone beta-1a.
ta-1a (infusione di placebo). L’incidenza di IRR è stata più elevata in assoluto durante Infezioni gravi sono state sviluppate dall’1,3% dei pazienti trattati con Ocrevus vs il
la Dose 1, infusione 1 (27,5%) per poi ridursi nel tempo fino al < 10% alla Dose 4. 2,9% dei pazienti trattati con interferone beta-1a. Nello studio controllato con pla-
Nella maggior parte dei casi, in entrambi i gruppi di trattamento le IRR hanno avuto cebo sulla SMPP si sono manifestate infezioni nel 72,2% dei pazienti trattati con
un’intensità da lieve a moderata. Il 21,7% e il 10,1% dei pazienti trattati con Ocrevus Ocrevus e nel 69,9% di quelli trattati con placebo. Infezioni gravi hanno interessato
hanno manifestato IRR rispettivamente lievi e moderate, il 2,4% ha manifestato IRR il 6,2% dei pazienti trattati con Ocrevus vs il 6,7% dei pazienti trattati con placebo.
severe e lo 0,1% IRR potenzialmente letali. Vedere paragrafo 4.4. Nella SMR è stato osservato un aumento del tasso di infezioni gravi tra gli anni 2 e
Nello studio clinico controllato con placebo (SMPP), le IRR hanno rappresentato l’evento 3, ma non in quelli successivi. Nella SMPP non è stato registrato alcun incremento.
avverso più comune nei pazienti trattati con Ocrevus, con un’incidenza complessiva del Infezioni delle vie respiratorie
40,1% rispetto a un’incidenza del 25,5% nel gruppo placebo. L’incidenza di IRR è stata La proporzione di infezioni delle vie respiratorie è stata più elevata tra i pazienti trat-
più elevata in assoluto durante la Dose 1, infusione 1 (27,4%,) per poi ridursi con le dosi tati con Ocrevus rispetto a quelli trattati con interferone beta-1a e placebo.
successive fino al < 10% alla Dose 4. Una proporzione maggiore di pazienti in ciascun Negli studi clinici sulla SMR, il 39,9% dei pazienti trattati con Ocrevus e il 33,2% dei
gruppo ha manifestato IRR con la prima infusione di ogni dose rispetto alla seconda pazienti trattati con interferone beta-1a ha manifestato un’infezione delle vie ae-
infusione della stessa dose. Nella maggior parte dei casi le IRR hanno avuto un’intensità ree superiori, mentre il 7,5% dei pazienti trattati con Ocrevus e il 5,2% dei pazienti
da lieve a moderata. Il 26,7% e l’11,9% dei pazienti trattati con Ocrevus hanno mani- trattati con interferone beta-1a ha manifestato un’infezione delle vie aeree inferiori.
festato IRR rispettivamente lievi e moderate; l’1,4% ha manifestato IRR severe. Non vi Nello studio clinico sulla SMPP, il 48,8% dei pazienti trattati con Ocrevus e il 42,7%
sono state IRR potenzialmente letali. Vedere paragrafo 4.4. dei pazienti trattati con placebo ha manifestato un’infezione delle vie aeree superiori,
L’alternativa dell’infusione in tempi ridotti per le dosi successive mentre il 9,9% dei pazienti trattati Ocrevus e il 9,2% dei pazienti trattati con placebo
In uno studio (sottostudio Shorter Infusion dello studio MA30143) volto a caratteriz- ha manifestato un’infezione delle vie aeree inferiori.
zare il profilo di sicurezza delle infusioni in tempi ridotti di Ocrevus (della durata di 2 Le infezioni delle vie aeree riferite in pazienti trattati con Ocrevus hanno avuto un’in-tensità prevalentemente da lieve a moderata (80-90%). somministrati in due infusioni endovenose da 1000 mg, a distanza di 2 settimane
Herpes l’una dall’altra (studio di fase II per la determinazione del dosaggio nella SMRR). Le
Negli studi clinici con controllo attivo (SMR), infezioni da herpes sono state riferite reazioni avverse al farmaco sono state in linea con il profilo di sicurezza di Ocrevus
con una frequenza maggiore tra i pazienti trattati con Ocrevus rispetto a quelli trattati emerso negli studi clinici registrativi.
con interferone-beta-1a e tra queste vi sono state infezioni da herpes zoster (2,1% Per informazioni sulla sindrome da risposta infiammatoria sistemica (SIRS) manife-
vs 1,0%), herpes simplex, (0,7 % vs 0,1%) herpes orale (3,0% vs 2,2%), herpes statasi in un paziente trattato con Ocrevus 2000 mg, vedere paragrafo 4.8.
genitale (0,1% vs 0%) ed herpes virus (0,1% vs 0%). Le infezioni hanno avuto una In caso di sovradosaggio non esiste un antidoto specifico; si deve interrompere im-
severità prevalentemente da lieve a moderata e i pazienti si sono ripresi ricorrendo mediatamente l’infusione e porre il paziente sotto osservazione per rilevare eventuali
a terapie standard. IRR (vedere paragrafo 4.4).
Nello studio clinico controllato con placebo (SMPP) è stata osservata una proporzione 5. PROPRIETÀ FARMACOLOGICHE
di pazienti con herpes orale maggiore (2,7% vs. 0,8%) nel braccio di trattamento 5.1 Proprietà farmacodinamiche
con Ocrevus. Categoria farmacoterapeutica: gruppo degli agenti immunosoppressori selettivi; co-
Esami di laboratorio dice ATC: L04AA36.
Immunoglobuline Meccanismo d’azione
Il trattamento con Ocrevus ha determinato una riduzione delle immunoglobuline totali Ocrelizumab è un anticorpo monoclonale ricombinante umanizzato che si lega selet-
nel periodo controllato degli studi, principalmente dovuta a una riduzione delle IgM. I tivamente alle cellule B esprimenti il CD20.
risultati degli studi clinici hanno mostrato un’associazione tra i livelli ridotti di IgG (e Il CD20 è un antigene di superficie presente sulle cellule pre-B, sulle cellule B mature
meno per IgM o IgA) e le infezioni gravi. e sulle cellule B della memoria, ma che non risulta espresso sulle cellule staminali
Linfociti linfoidi e sulle plasmacellule.
Nella SMR è stata osservata una riduzione dei livelli di linfociti < LLN nel 20,7% dei I meccanismi precisi con cui ocrelizumab esercita i suoi effetti clinici terapeutici
pazienti trattati con Ocrevus e nel 32,6% di quelli trattati con interferone beta-1a. nella SM non sono stati del tutto chiariti, ma si suppone che implichino l’immu-
Con la SMPP è stata osservata una riduzione dei livelli di linfociti < LLN nel 26,3% dei nomodulazione attraverso la riduzione del numero e della funzione delle cellule B
pazienti trattati con Ocrevus e nell’11,7% di quelli trattati con placebo. esprimenti CD20. Una volta legatosi alla superficie cellulare, ocrelizumab determina
La maggior parte di queste riduzioni segnalate nei pazienti trattati con Ocrevus è una deplezione selettiva delle cellule B esprimenti CD20 mediante fagocitosi cellu-
stata di severità di grado 1 (< LLN - 800 cellule/mm3) e 2 (tra 500 e 800 cellule/mm3). lare anticorpo-mediata (Antibody-Dependent Cellular Phagocytosis, ADCP), citotossi-
Circa l’1% dei pazienti del gruppo Ocrevus ha manifestato linfopenia di grado 3 (tra cità cellulomediata anticorpo-dipendente (Antibody-Dependent Cellular Cytotoxicity,
200 e 500 cellule/mm3). Nessuno dei pazienti ha manifestato linfopenia di grado 4 ADCC), citotossicità complemento-dipendente (Complement-Dependent Cytotoxicity,
(< 200 cellule/mm3). CDC) e apoptosi. La capacità di ricostituzione delle cellule B e l’immunità umorale
Nei pazienti trattati con ocrelizumab, durante episodi di diminuzione confermata del- preesistente sono preservate. Inoltre, l’immunità innata e la conta totale delle cellule
la conta linfocitaria totale, è stato osservato un aumento del tasso di infezioni gravi. Il T non sono interessati da questa azione.
numero di infezioni gravi era troppo basso per trarre conclusioni definitive. Effetti farmacodinamici
Neutrofili Il trattamento con Ocrevus determina una rapida deplezione delle cellule B CD19+
Nel periodo di trattamento con controllo attivo (SMR) è stata osservata una riduzione nel sangue entro 14 giorni dal trattamento (primo punto temporale di valutazione)
dei neutrofili < LNN nel 14,7% dei pazienti trattati con Ocrevus rispetto al 40,9% come effetto farmacologico atteso. Questo effetto è risultato sostenuto per l’intera
dei pazienti trattati con interferone beta-1a. Nello studio clinico controllato con pla- durata del periodo di trattamento. Per i conteggi delle cellule B si utilizza il CD19
cebo (SMPP), la proporzione di pazienti trattati con Ocrevus che ha fatto osservare poiché la presenza di Ocrevus interferisce con il riconoscimento di CD20 mediante
una riduzione dei neutrofili è stata superiore (12,9%) rispetto ai pazienti trattati con saggio.
placebo (10,0%). Tra questi, una percentuale superiore di pazienti (4,3%) nel gruppo Negli studi di fase III, tra una dose di Ocrevus e l’altra, fino al 5% dei pazienti ha
Ocrevus ha manifestato neutropenia di grado uguale o superiore a 2, contro l’1,3% mostrato ricostituzione cellule B (> limite inferiore della norma (LLN) o valore basale)
registrato nel gruppo placebo; circa l’1% dei pazienti del gruppo Ocrevus ha manife- in almeno un punto temporale. L’entità e la durata della deplezione delle cellule B è
stato neutropenia di grado 4, contro lo 0% registrato nel gruppo placebo. stata coerente negli studi sulla SMPP e sulla SMR.
Nella maggior parte dei casi le riduzioni dei neutrofili sono state transitorie (osservate Il follow-up più lungo effettuato dopo l’ultima infusione di Ocrevus (studio di fase II
soltanto una volta per un dato paziente trattato con Ocrevus) e di grado 1 (< 1500 WA21493, N = 51) indica che la mediana del tempo di ricostituzione delle cellule B
cellule/mm3) e 2 (tra 1000 e 1500 cellule/mm3) di severità. Un paziente con neutropenia (ritorno al basale/LLN, a seconda del caso che si verifica per primo) è stata di 72 set-
di grado 3 (tra 500 e 1000 cellule/mm3) e un paziente con neutropenia di grado 4 (< timane (range: 27-175 settimane). Il 90% di tutti i pazienti ha fatto osservare una
500 cellule/mm3) hanno avuto bisogno di trattamento specifico con fattore stimolante ricostituzione delle cellule B al valore LLN o al basale entro circa due anni e mezzo
le colonie granulocitarie e hanno proseguito la terapia con ocrelizumab dopo l’episodio. dopo l’ultima infusione.
Altro Efficacia e sicurezza clinica
Un paziente trattato con 2000 mg di Ocrevus è deceduto a causa di sindrome da Forme recidivanti di SM
risposta infiammatoria sistemica (SIRS) di eziologia ignota a seguito di una risonanza L’efficacia e la sicurezza di Ocrevus sono state valutate in due studi clinici ran-
magnetica (RM) 12 settimane dopo l’ultima infusione; una reazione anafilattoide al domizzati di disegno identico (WA21092 e WA21093), in doppio cieco, con doppia
mezzo di contrasto contenente gadolinio usato nella RM potrebbe aver contribuito simulazione, con controllo attivo, condotti in pazienti con forme recidivanti di SM
allo sviluppo della SIRS. (ai sensi dei criteri McDonald 2010) ed evidenza di attività di malattia (in base
Segnalazione delle reazioni avverse sospette alle caratteristiche cliniche o radiologiche) nei due anni precedenti. Disegno del-
La segnalazione delle reazioni avverse sospette che si verificano dopo l’autorizza- lo studio e caratteristiche basali della popolazione in studio sono riassunti nella
zione del medicinale è importante, in quanto permette un monitoraggio continuo Tabella 3.
del rapporto beneficio/rischio del medicinale. Agli operatori sanitari è richiesto Le caratteristiche demografiche e basali sono risultate ben bilanciate tra i due gruppi
di segnalare qualsiasi reazione avversa sospetta tramite il sistema nazionale di di trattamento. I pazienti trattati con Ocrevus (Gruppo A) hanno ricevuto 600 mg ogni
segnalazione riportato all’indirizzo http://www.aifa.gov.it/content/segnalazioni-re- 6 mesi (Dose 1 in forma di 2 infusioni endovenose da 300 mg, somministrate a
azioni-avverse. distanza di 2 settimane l’una dall’altra, e dosi successive somministrate mediante
4.9 Sovradosaggio singola infusione endovenosa da 600 mg). I pazienti del Gruppo B hanno ricevu-
Vi è una limitata esperienza clinica con dosi superiori rispetto alla dose endovenosa to interferone beta-1a (Rebif) alla dose di 44 mcg mediante iniezione sottocutanea
approvata di Ocrevus. La dose più alta testata ad oggi in pazienti con SM è 2000 mg, 3 volte a settimana.
Tabella 3. Disegno dello studio, caratteristiche basali e demografiche
Studio 1 Studio 2
Nome dello studio WA21092 (OPERA I) WA21093 (OPERA II)
(n = 821) (n = 835)
Disegno dello studio
Popolazione in studio Pazienti con forme recidivanti di SM
Storia della malattia allo screening Almeno due recidive nei due anni precedenti o una recidiva nell’anno precedente;
punteggio EDSS* compreso tra 0 e 5,5 incluso
Durata dello studio 2 anni
Gruppi di trattamento Gruppo A: Ocrevus 600 mg
Gruppo B: interferone beta-1a 44 mcg SC (IFN)
Caratteristiche basali Ocrevus IFN Ocrevus IFN
600 mg 44 mcg 600 mg 44 mcg
(n = 410) (n = 411) (n = 417) (n = 418)
Età media (anni) 37,1 36,9 37,2 37,4Puoi anche leggere

















































